

Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth
- PMID: 23034650
- PMCID: PMC3601659
- DOI: 10.1038/nature11465
Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth
Abstract
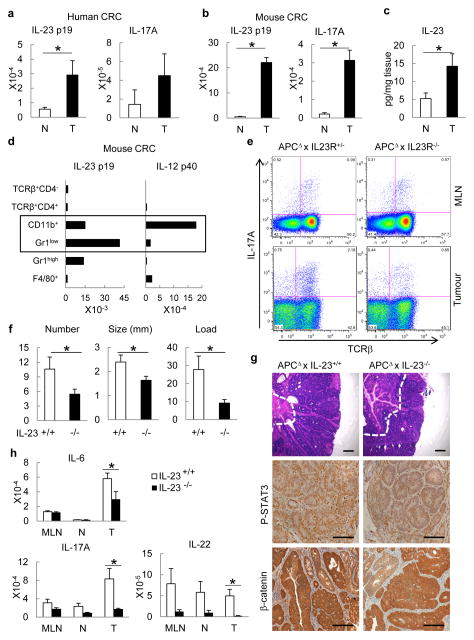
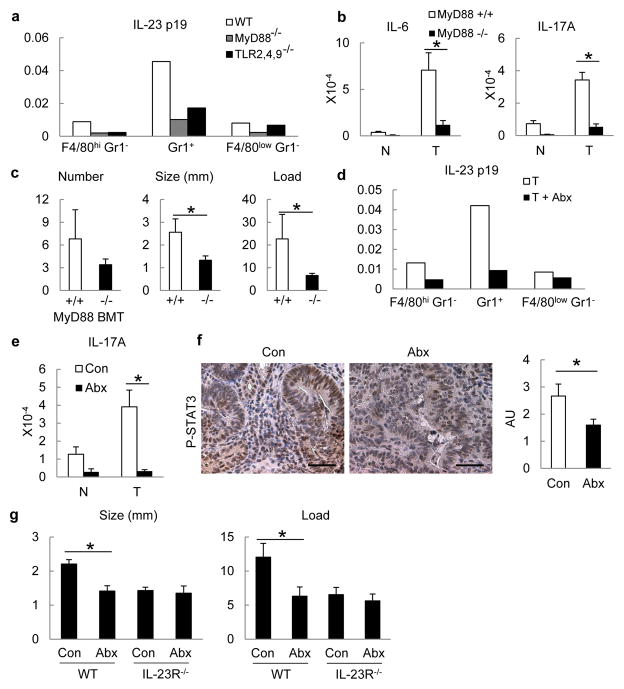
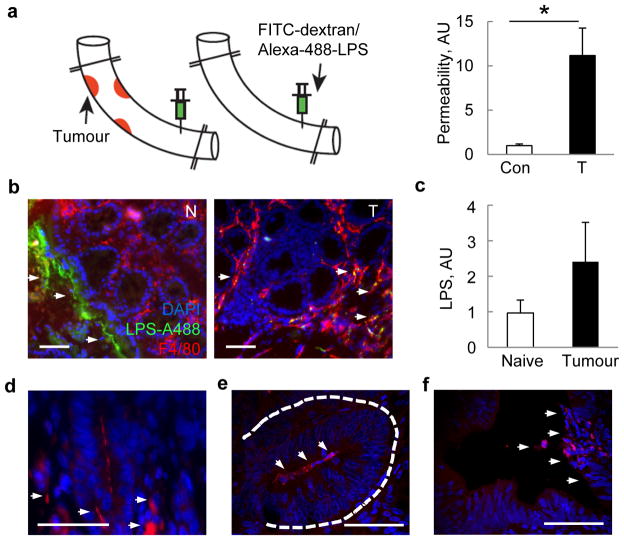
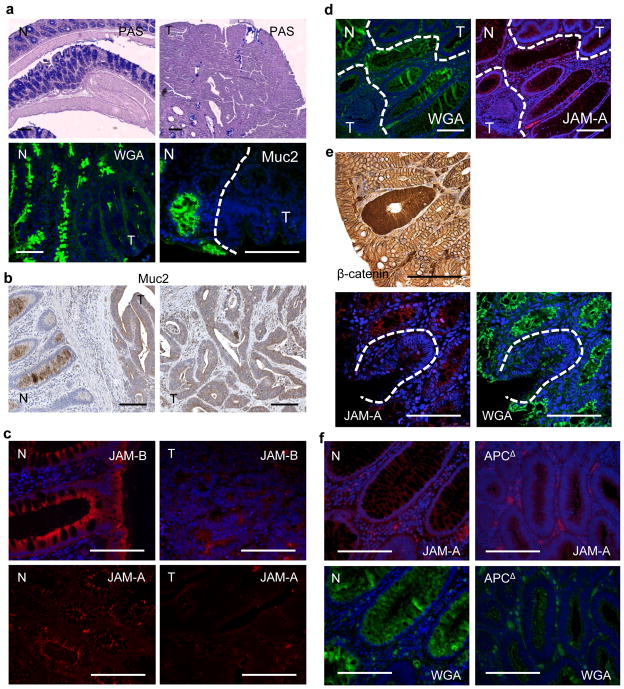
Approximately 2% of colorectal cancer is linked to pre-existing inflammation known as colitis-associated cancer, but most develops in patients without underlying inflammatory bowel disease. Colorectal cancer often follows a genetic pathway whereby loss of the adenomatous polyposis coli (APC) tumour suppressor and activation of β-catenin are followed by mutations in K-Ras, PIK3CA and TP53, as the tumour emerges and progresses. Curiously, however, 'inflammatory signature' genes characteristic of colitis-associated cancer are also upregulated in colorectal cancer. Further, like most solid tumours, colorectal cancer exhibits immune/inflammatory infiltrates, referred to as 'tumour-elicited inflammation'. Although infiltrating CD4(+) T(H)1 cells and CD8(+) cytotoxic T cells constitute a positive prognostic sign in colorectal cancer, myeloid cells and T-helper interleukin (IL)-17-producing (T(H)17) cells promote tumorigenesis, and a 'T(H)17 expression signature' in stage I/II colorectal cancer is associated with a drastic decrease in disease-free survival. Despite its pathogenic importance, the mechanisms responsible for the appearance of tumour-elicited inflammation are poorly understood. Many epithelial cancers develop proximally to microbial communities, which are physically separated from immune cells by an epithelial barrier. We investigated mechanisms responsible for tumour-elicited inflammation in a mouse model of colorectal tumorigenesis, which, like human colorectal cancer, exhibits upregulation of IL-23 and IL-17. Here we show that IL-23 signalling promotes tumour growth and progression, and development of a tumoural IL-17 response. IL-23 is mainly produced by tumour-associated myeloid cells that are likely to be activated by microbial products, which penetrate the tumours but not adjacent tissue. Both early and late colorectal neoplasms exhibit defective expression of several barrier proteins. We propose that barrier deterioration induced by colorectal-cancer-initiating genetic lesions results in adenoma invasion by microbial products that trigger tumour-elicited inflammation, which in turn drives tumour growth.
Conflict of interest statement
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
Figures
References
-
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature Med. 2004;10:789–799. - PubMed
-
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. - PubMed
-
- Wood LD, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. - PubMed
-
- Reichling T, et al. Transcriptional profiles of intestinal tumors in Apc(Min) mice are unique from those of embryonic intestine and identify novel gene targets dysregulated in human colorectal tumors. Cancer Res. 2005;65:166–176. - PubMed
-
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 AA020703/AA/NIAAA NIH HHS/United States
- AI043477/AI/NIAID NIH HHS/United States
- R24 DK080506/DK/NIDDK NIH HHS/United States
- K99 DK088589/DK/NIDDK NIH HHS/United States
- K99-DK088589/DK/NIDDK NIH HHS/United States
- DK035108/DK/NIDDK NIH HHS/United States
- K08 DK081830/DK/NIDDK NIH HHS/United States
- R01 AI043477/AI/NIAID NIH HHS/United States
- R01 AI050265/AI/NIAID NIH HHS/United States
- DK080506/DK/NIDDK NIH HHS/United States
- P01 DK035108/DK/NIDDK NIH HHS/United States
- R37 AI043477/AI/NIAID NIH HHS/United States
- R01 CA082223/CA/NCI NIH HHS/United States
- R01CA082223/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
