

Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains
- PMID: 23034917
- PMCID: PMC3638323
- DOI: 10.1002/ana.23614
Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains
Abstract
Objective: Mutations in the glucocerebrosidase gene (GBA) represent a significant risk factor for developing Parkinson disease (PD). We investigated the enzymatic activity of glucocerebrosidase (GCase) in PD brains carrying heterozygote GBA mutations (PD+GBA) and sporadic PD brains.
Methods: GCase activity was measured using a fluorescent assay in cerebellum, frontal cortex, putamen, amygdala, and substantia nigra of PD+GBA (n = 9-14) and sporadic PD brains (n = 12-14). Protein expression of GCase and other lysosomal proteins was determined by western blotting. The relation between GCase, α-synuclein, and mitochondria function was also investigated in vitro.
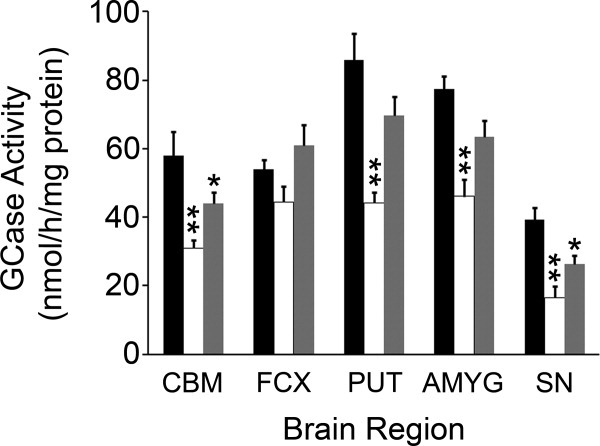
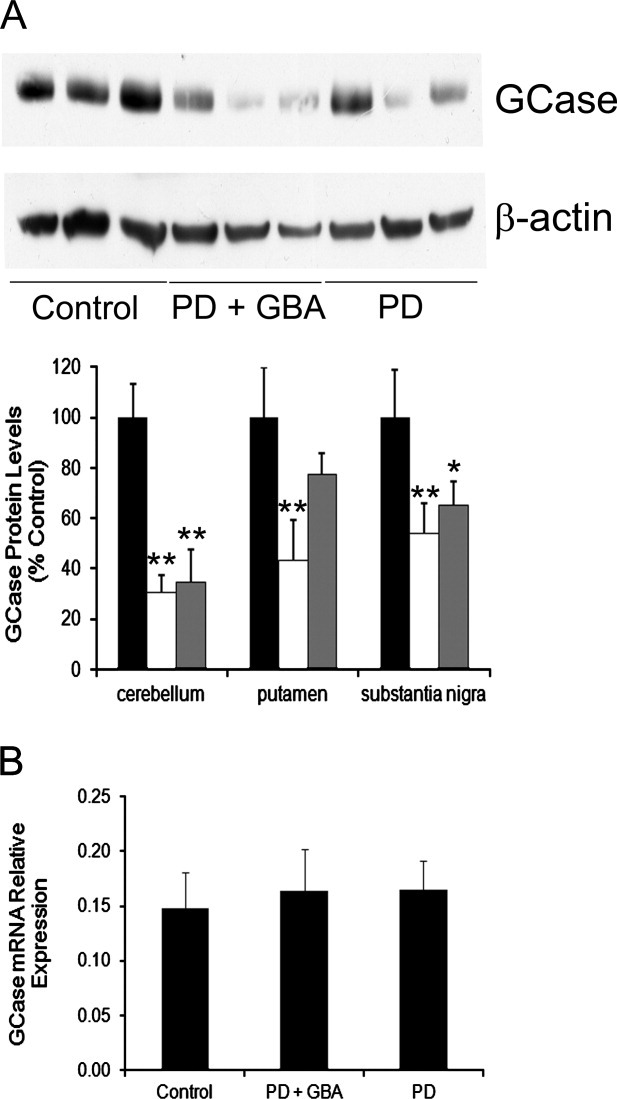
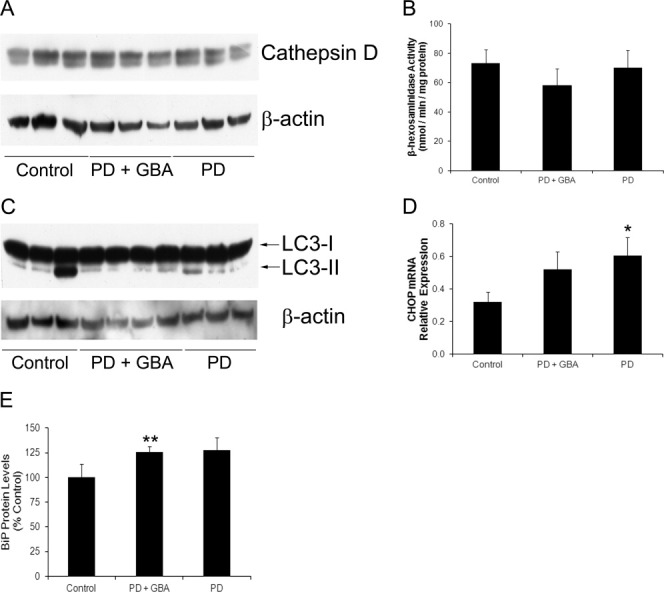
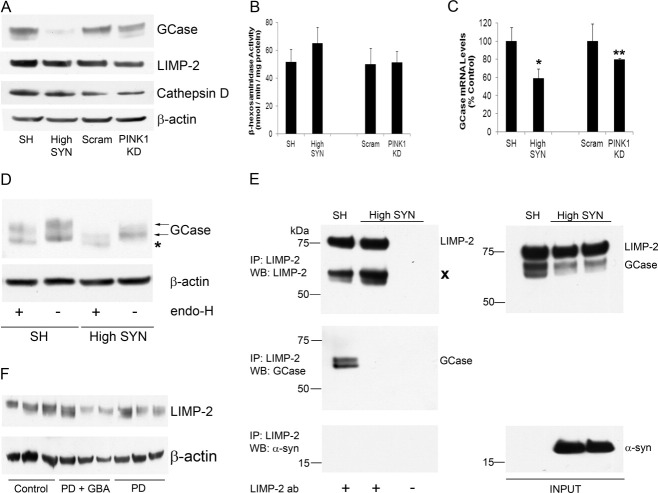
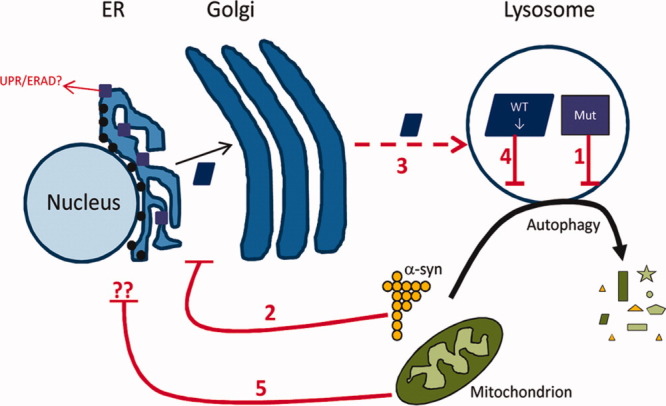
Results: A significant decrease in GCase activity was observed in all PD+GBA brain areas except the frontal cortex. The greatest deficiency was in the substantia nigra (58% decrease; p < 0.01). GCase activity was also significantly decreased in the substantia nigra (33% decrease; p < 0.05) and cerebellum (24% decrease; p < 0.05) of sporadic PD brains. GCase protein expression was lower in PD+GBA and PD brains, whereas increased C/EBP homologous protein and binding immunoglobulin protein levels indicated that the unfolded protein response was activated. Elevated α-synuclein levels or PTEN-induced putative kinase 1 deficiency in cultured cells had a significant effect on GCase protein levels.
Interpretation: GCase deficiency in PD brains with GBA mutations is a combination of decreased catalytic activity and reduced protein levels. This is most pronounced in the substantia nigra. Biochemical changes involved in PD pathogenesis affect wild-type GCase protein expression in vitro, and these could be contributing factors to the GCase deficiency observed in sporadic PD brains.
Copyright © 2012 American Neurological Association.
Figures
References
-
- Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet. 2008;372:1263–1271. - PubMed
-
- Schapira AHV. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97–109. - PubMed
-
- Neudorfer O, Giladi N, Elstein D, et al. Occurrence of Parkinson's syndrome in type I Gaucher disease. QJM. 1996;89:691–694. - PubMed
-
- Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med. 2004;351:1972–1977. - PubMed
-
- Wong K, Sidransky E, Verma A, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82:192–207. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
