

Tissue specificity of a human mitochondrial disease: differentiation-enhanced mis-splicing of the Fe-S scaffold gene ISCU renders patient cells more sensitive to oxidative stress in ISCU myopathy
- PMID: 23035118
- PMCID: PMC3504726
- DOI: 10.1074/jbc.M112.418889
Tissue specificity of a human mitochondrial disease: differentiation-enhanced mis-splicing of the Fe-S scaffold gene ISCU renders patient cells more sensitive to oxidative stress in ISCU myopathy
Abstract
Background: ISCU myopathy is a disease caused by muscle-specific deficiency of the Fe-S cluster scaffold protein ISCU.
Results: MyoD expression enhanced ISCU mRNA mis-splicing, and oxidative stress exacerbated ISCU depletion in patient cells.
Conclusion: ISCU protein deficiency in patients results from muscle-specific mis-splicing as well as oxidative stress.
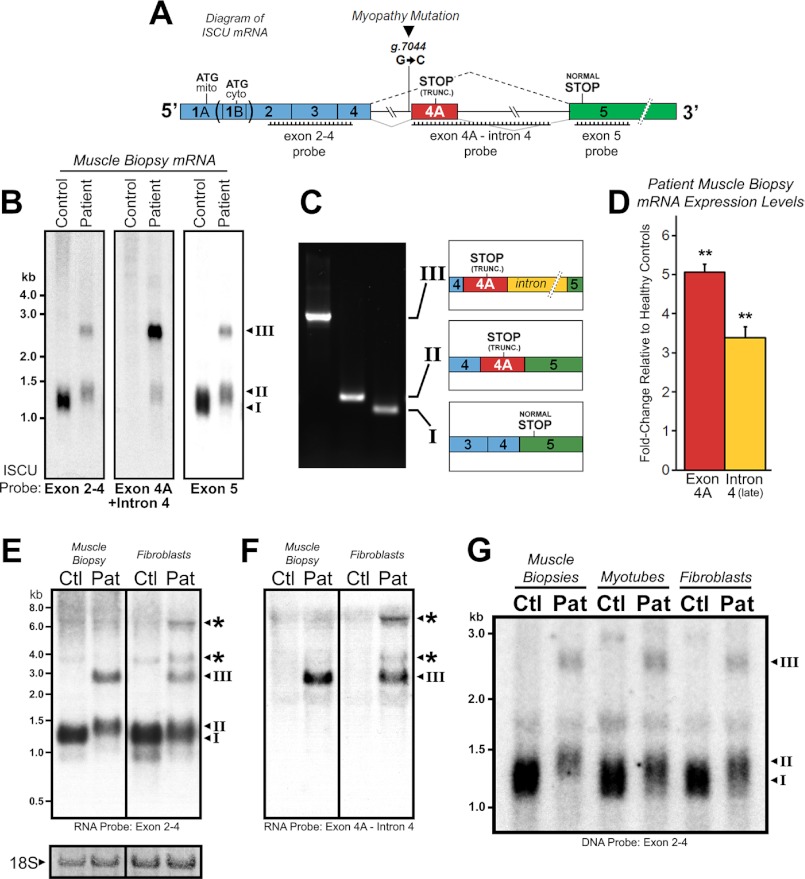
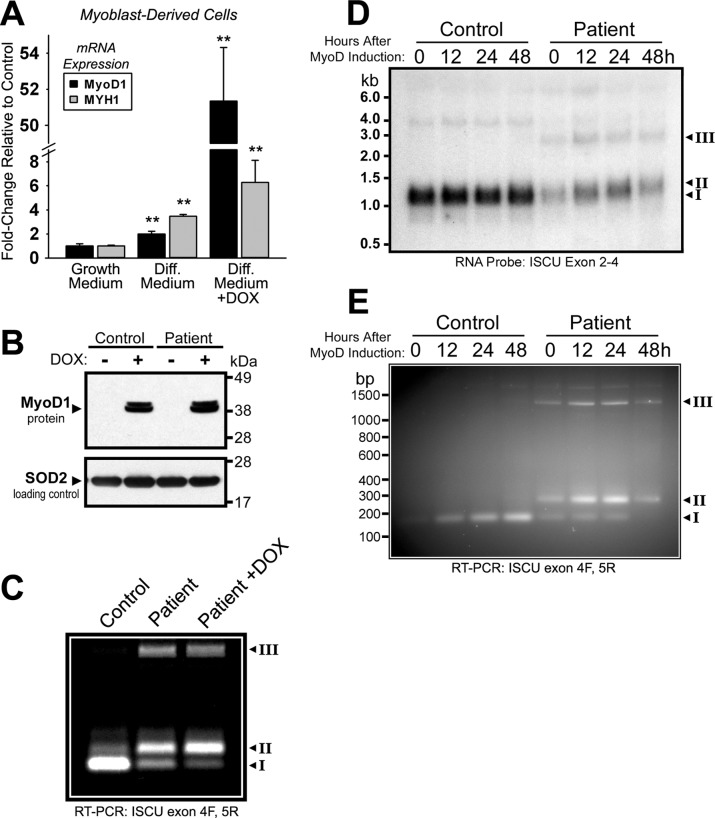
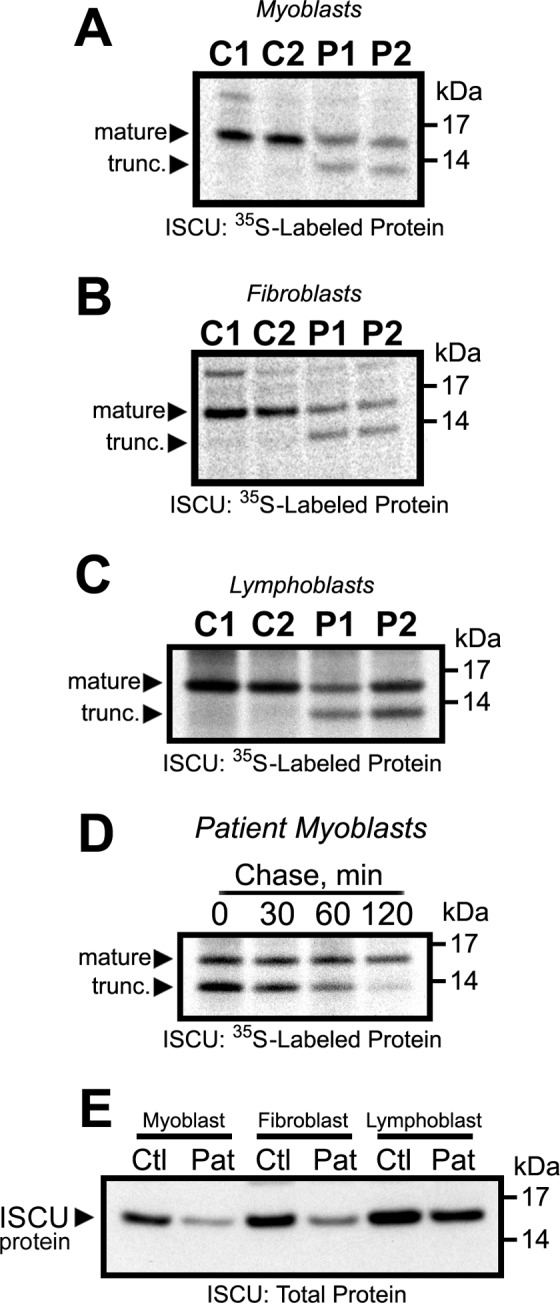
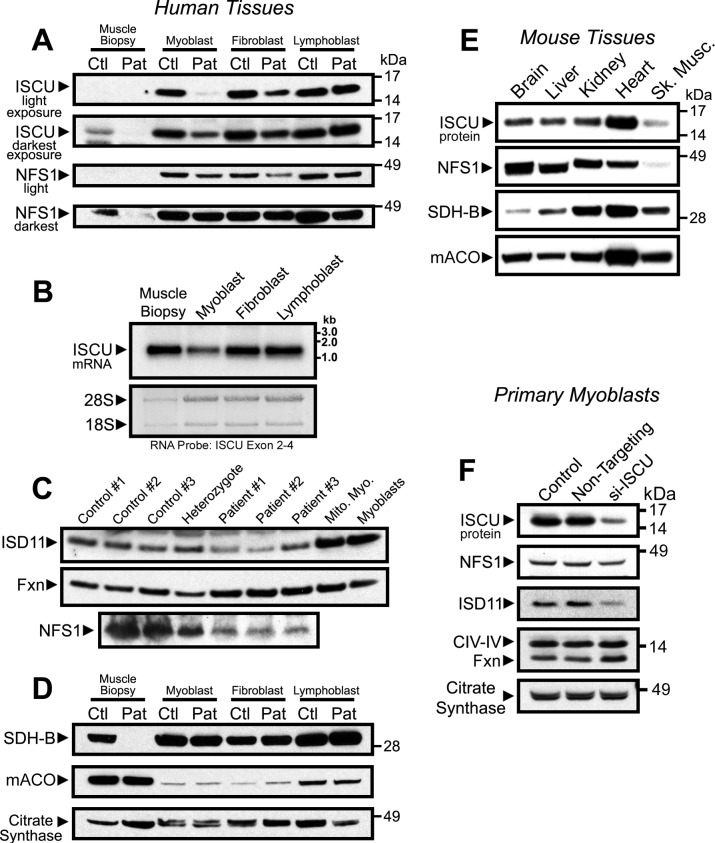
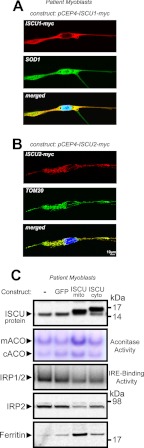
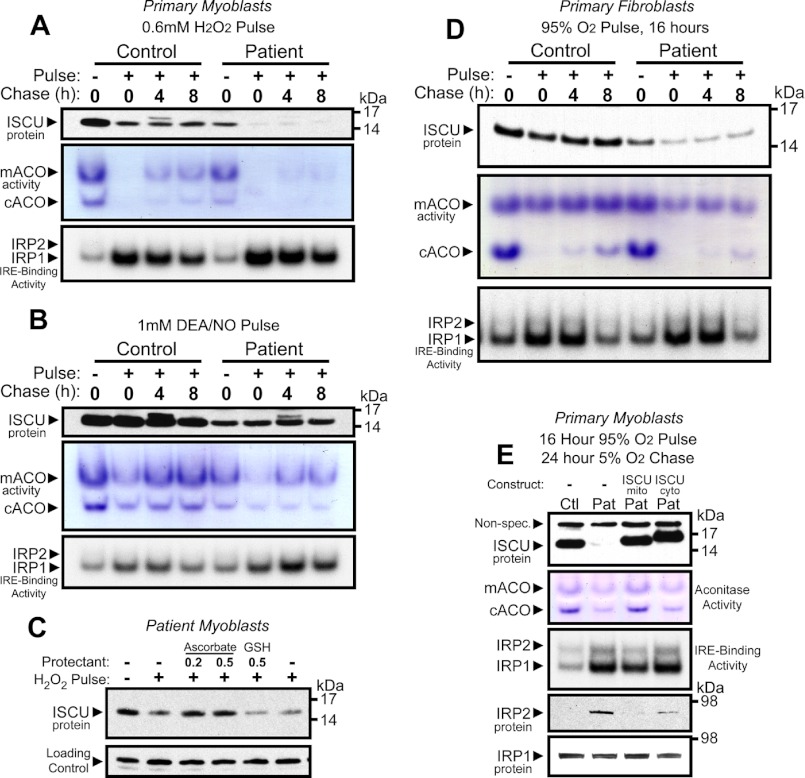
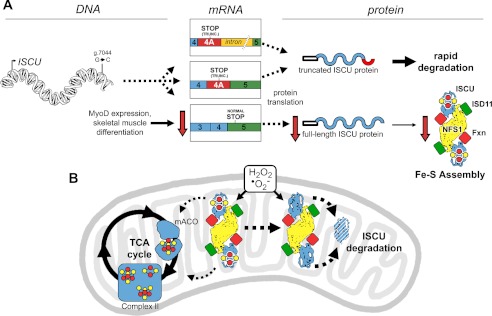
Significance: Oxidative stress negatively influences the mammalian Fe-S cluster assembly machinery by destabilization of ISCU. Iron-sulfur (Fe-S) cluster cofactors are formed on the scaffold protein ISCU. ISCU myopathy is a disease caused by an intronic mutation that leads to abnormally spliced ISCU mRNA. We found that two predominant mis-spliced ISCU mRNAs produce a truncated and short-lived ISCU protein product in multiple patient cell types. Expression of the muscle-specific transcription factor MyoD further diminished normal splicing of ISCU mRNA in patient myoblasts, demonstrating that the process of muscle differentiation enhances the loss of normal ISCU mRNA splicing. ISCU protein was nearly undetectable in patient skeletal muscle, but was higher in patient myoblasts, fibroblasts, and lymphoblasts. We next treated patient cells with pro-oxidants to mimic the oxidative stress associated with muscle activity. Brief hydrogen peroxide treatment or incubation in an enriched oxygen atmosphere led to a marked further reduction of ISCU protein levels, which could be prevented by pretreatment with the antioxidant ascorbate. Thus, we conclude that skeletal muscle differentiation of patient cells causes a higher degree of abnormal ISCU splicing and that oxidative stress resulting from skeletal muscle work destabilizes the small amounts of normal ISCU protein generated in patient skeletal muscles.
Figures
Similar articles
-
Tissue-specific splicing of ISCU results in a skeletal muscle phenotype in myopathy with lactic acidosis, while complete loss of ISCU results in early embryonic death in mice.Hum Genet. 2011 Apr;129(4):371-8. doi: 10.1007/s00439-010-0931-3. Epub 2010 Dec 17. Hum Genet. 2011. PMID: 21165651
-
Differences in RNA processing underlie the tissue specific phenotype of ISCU myopathy.Biochim Biophys Acta. 2010 Jun;1802(6):539-44. doi: 10.1016/j.bbadis.2010.02.010. Epub 2010 Mar 4. Biochim Biophys Acta. 2010. PMID: 20206689
-
The High Level of Aberrant Splicing of ISCU in Slow-Twitch Muscle May Involve the Splicing Factor SRSF3.PLoS One. 2016 Oct 26;11(10):e0165453. doi: 10.1371/journal.pone.0165453. eCollection 2016. PLoS One. 2016. PMID: 27783661 Free PMC article.
-
Mitochondrial iron-sulfur protein biogenesis and human disease.Biochimie. 2014 May;100:61-77. doi: 10.1016/j.biochi.2014.01.010. Epub 2014 Jan 23. Biochimie. 2014. PMID: 24462711 Review.
-
Mammalian Fe-S cluster biogenesis and its implication in disease.Biochimie. 2014 May;100:48-60. doi: 10.1016/j.biochi.2014.01.009. Epub 2014 Jan 17. Biochimie. 2014. PMID: 24440636 Review.
Cited by
-
Differential diagnosis of lipoic acid synthesis defects.J Inherit Metab Dis. 2016 Nov;39(6):781-793. doi: 10.1007/s10545-016-9975-4. Epub 2016 Sep 1. J Inherit Metab Dis. 2016. PMID: 27586888 Review.
-
Complete genome analysis of sugarcane root associated endophytic diazotroph Pseudomonas aeruginosa DJ06 revealing versatile molecular mechanism involved in sugarcane development.Front Microbiol. 2023 Apr 20;14:1096754. doi: 10.3389/fmicb.2023.1096754. eCollection 2023. Front Microbiol. 2023. PMID: 37152763 Free PMC article.
-
NMR as a Tool to Investigate the Processes of Mitochondrial and Cytosolic Iron-Sulfur Cluster Biosynthesis.Molecules. 2018 Aug 31;23(9):2213. doi: 10.3390/molecules23092213. Molecules. 2018. PMID: 30200358 Free PMC article. Review.
-
Function and crystal structure of the dimeric P-loop ATPase CFD1 coordinating an exposed [4Fe-4S] cluster for transfer to apoproteins.Proc Natl Acad Sci U S A. 2018 Sep 25;115(39):E9085-E9094. doi: 10.1073/pnas.1807762115. Epub 2018 Sep 10. Proc Natl Acad Sci U S A. 2018. PMID: 30201724 Free PMC article.
-
Protein-mediated assembly of succinate dehydrogenase and its cofactors.Crit Rev Biochem Mol Biol. 2015 Mar-Apr;50(2):168-80. doi: 10.3109/10409238.2014.990556. Epub 2014 Dec 9. Crit Rev Biochem Mol Biol. 2015. PMID: 25488574 Free PMC article. Review.
References
-
- Linderholm H., Müller R., Ringqvist T., Sörnäs R. (1969) Hereditary abnormal muscle metabolism with hyperkinetic circulation during exercise. Acta Med. Scand. 185, 153–166 - PubMed
-
- Kollberg G., Melberg A., Holme E., Oldfors A. (2011) Transient restoration of succinate dehydrogenase activity after rhabdomyolysis in iron-sulphur cluster deficiency myopathy. Neuromuscul. Disord. 21, 115–120 - PubMed
-
- Haller R. G., Henriksson K. G., Jorfeldt L., Hultman E., Wibom R., Sahlin K., Areskog N. H., Gunder M., Ayyad K., Blomqvist C. G., et al. (1991) Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J. Clin. Invest. 88, 1197–1206 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
