

Population genetic inference from personal genome data: impact of ancestry and admixture on human genomic variation
- PMID: 23040495
- PMCID: PMC3484644
- DOI: 10.1016/j.ajhg.2012.08.025
Population genetic inference from personal genome data: impact of ancestry and admixture on human genomic variation
Abstract
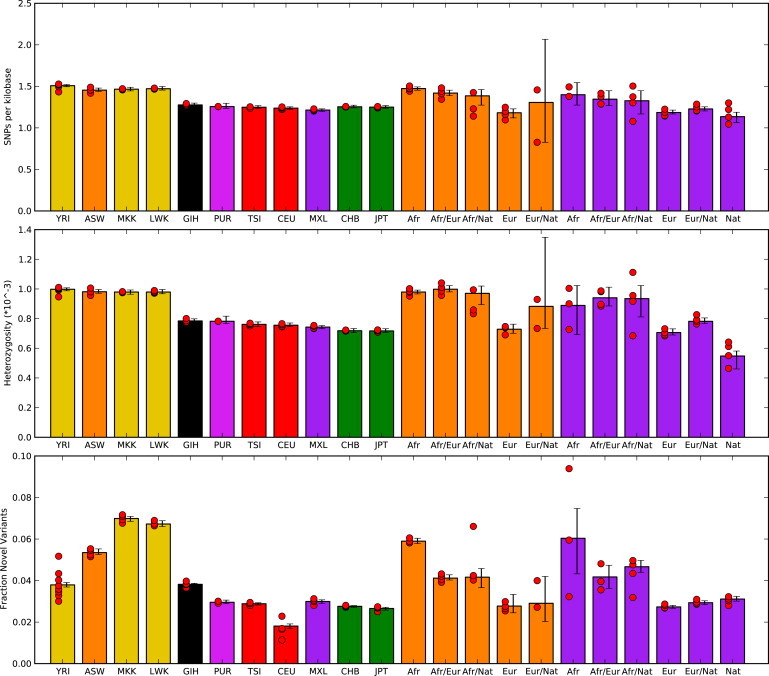
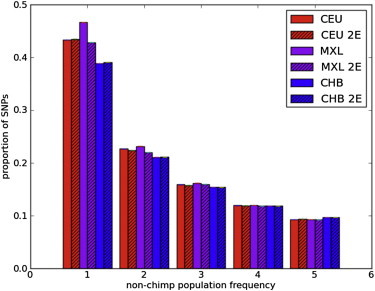
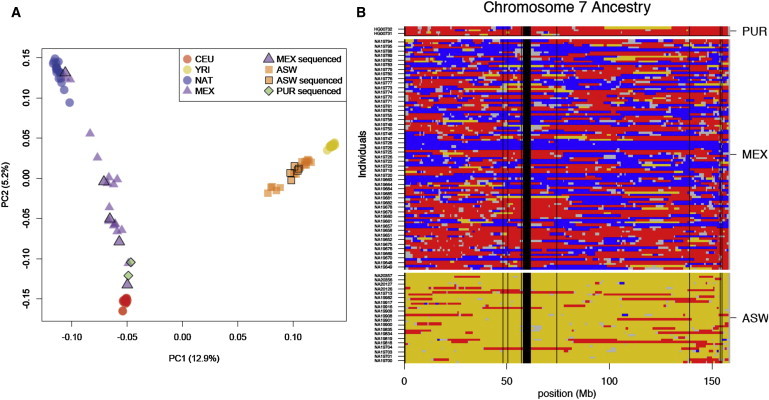
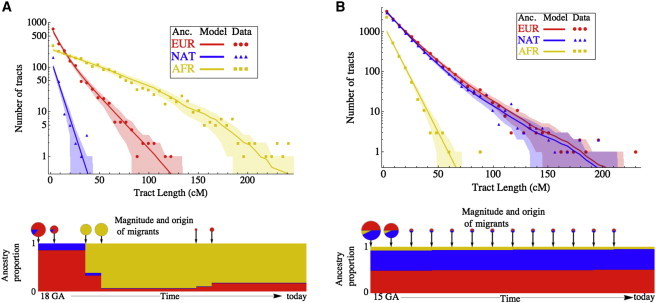
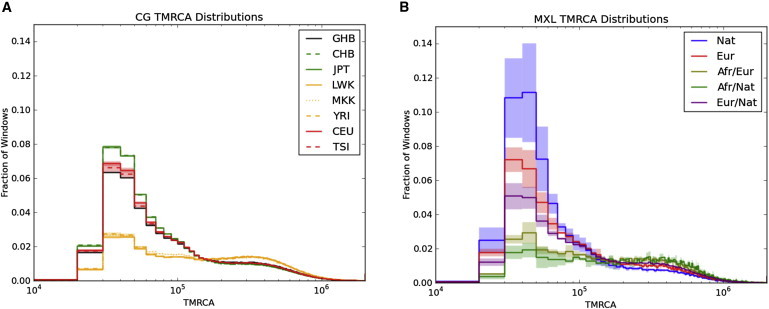
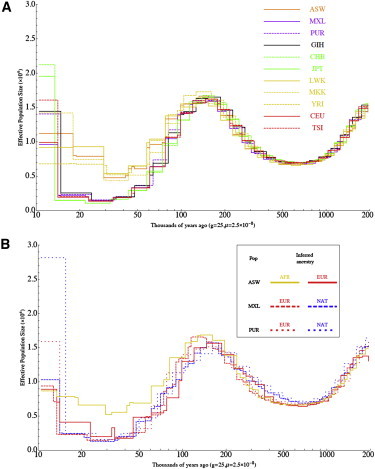
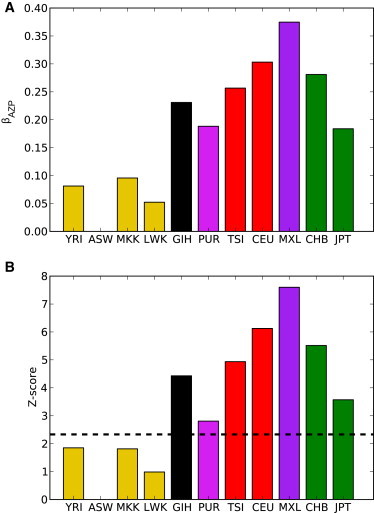
Full sequencing of individual human genomes has greatly expanded our understanding of human genetic variation and population history. Here, we present a systematic analysis of 50 human genomes from 11 diverse global populations sequenced at high coverage. Our sample includes 12 individuals who have admixed ancestry and who have varying degrees of recent (within the last 500 years) African, Native American, and European ancestry. We found over 21 million single-nucleotide variants that contribute to a 1.75-fold range in nucleotide heterozygosity across diverse human genomes. This heterozygosity ranged from a high of one heterozygous site per kilobase in west African genomes to a low of 0.57 heterozygous sites per kilobase in segments inferred to have diploid Native American ancestry from the genomes of Mexican and Puerto Rican individuals. We show evidence of all three continental ancestries in the genomes of Mexican, Puerto Rican, and African American populations, and the genome-wide statistics are highly consistent across individuals from a population once ancestry proportions have been accounted for. Using a generalized linear model, we identified subtle variations across populations in the proportion of neutral versus deleterious variation and found that genome-wide statistics vary in admixed populations even once ancestry proportions have been factored in. We further infer that multiple periods of gene flow shaped the diversity of admixed populations in the Americas-70% of the European ancestry in today's African Americans dates back to European gene flow happening only 7-8 generations ago.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
