

Genetics of hearing and deafness
- PMID: 23044516
- PMCID: PMC4523052
- DOI: 10.1002/ar.22579
Genetics of hearing and deafness
Erratum in
-
Corrigendum.Anat Rec (Hoboken). 2015 Nov;298(11):1815. doi: 10.1002/ar.23260. Anat Rec (Hoboken). 2015. PMID: 26463460 No abstract available.
Abstract
This article is a review of the genes and genetic disorders that affect hearing in humans and a few selected mouse models of deafness. Genetics is playing an increasingly critical role in the practice of medicine. This is not only in part to the importance that genetic knowledge has on traditional genetic diseases but also in part to the fact that genetic knowledge provides an understanding of the fundamental biological process of most diseases. The proteins coded by the genes related to hearing loss (HL) are involved in many functions in the ear, such as cochlear fluid homeostasis, ionic channels, stereocilia morphology and function, synaptic transmission, gene regulation, and others. Mouse models play a crucial role in understanding of the pathogenesis associated with these genes. Different types of familial HL have been recognized for years; however, in the last two decades, there has been tremendous progress in the discovery of gene mutations that cause deafness. Most of the cases of genetic deafness recognized today are monogenic disorders that can be broadly classified by the mode of inheritance (i.e., autosomal dominant, autosomal recessive, X-linked, and mitochondrial inheritance) and by the presence of associated phenotypic features (i.e., syndromic; and nonsyndromic). In terms of nonsyndromic HL, the chromosomal locations are currently known for ∼ 125 loci (54 for dominant and 71 for recessive deafness), 64 genes have been identified (24 for dominant and 40 for recessive deafness), and there are many more loci for syndromic deafness and X-linked and mitochondrial DNA disorders (http://hereditaryhearingloss.org). Thus, today's clinician must understand the science of medical genetics as this knowledge can lead to more effective disease diagnosis, counseling, treatment, and prevention.
Copyright © 2012 Wiley Periodicals, Inc.
Figures

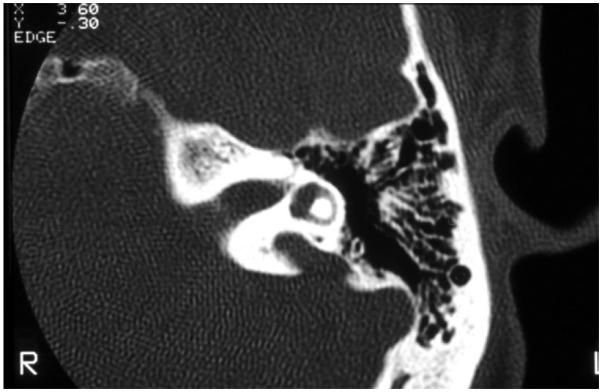
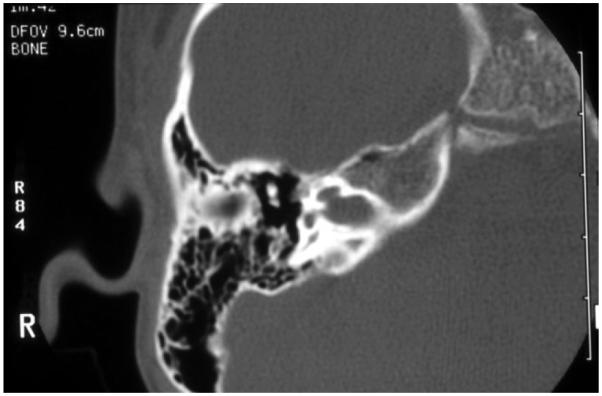
References
-
- Adato A, Michel V, Kikkawa Y, Reiners J, Alagramam KN, Weil D, Yonekawa H, Wolfrum U, El-Amraoui A, Petit C. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14:347–356. - PubMed
-
- Avraham KB. Mouse models for deafness: lessons for the human inner ear and hearing loss. Ear Hear. 2003;24:332–341. - PubMed
-
- Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. NatGenet. 1995;11:369–375. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
