

Evolution of the cancer genome
- PMID: 23044827
- PMCID: PMC3666082
- DOI: 10.1038/nrg3317
Evolution of the cancer genome
Abstract
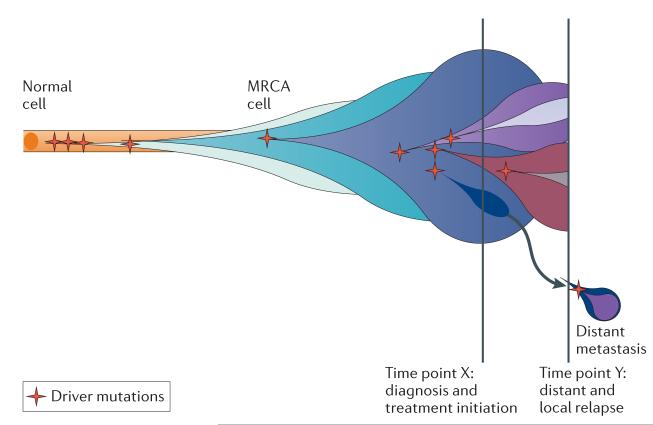
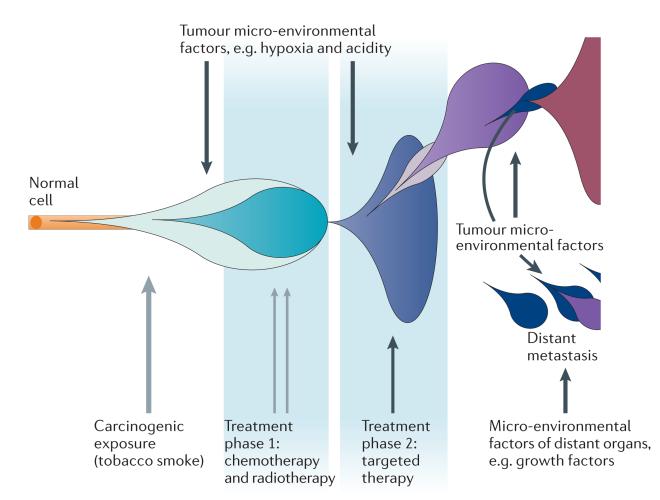
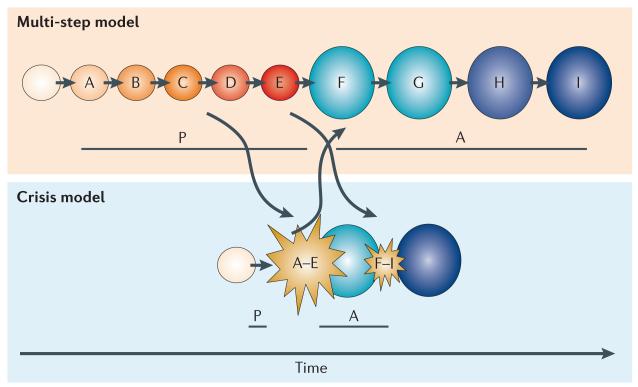
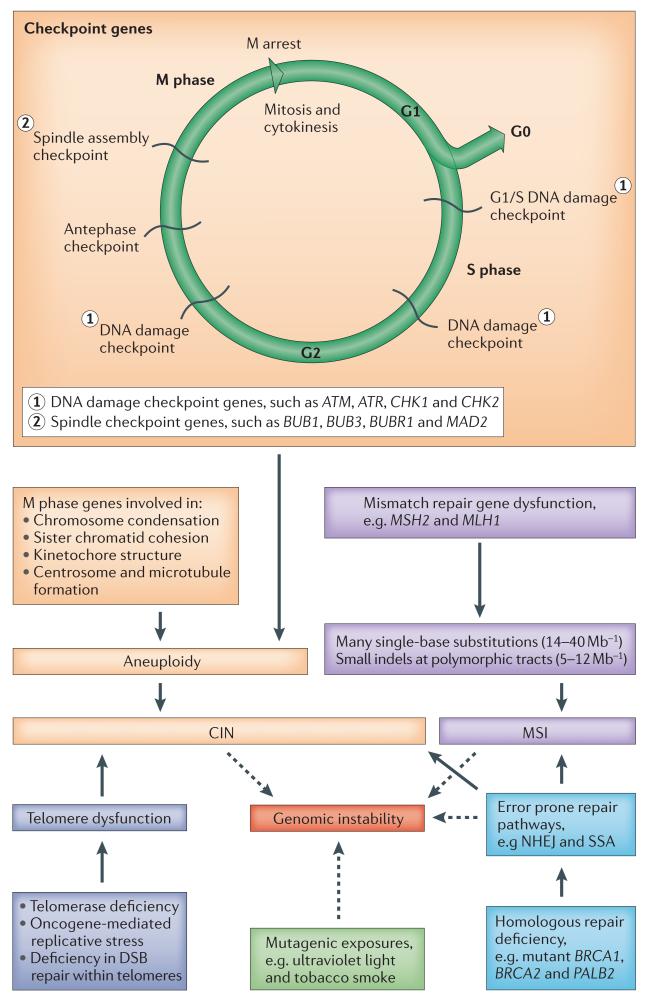
The advent of massively parallel sequencing technologies has allowed the characterization of cancer genomes at an unprecedented resolution. Investigation of the mutational landscape of tumours is providing new insights into cancer genome evolution, laying bare the interplay of somatic mutation, adaptation of clones to their environment and natural selection. These studies have demonstrated the extent of the heterogeneity of cancer genomes, have allowed inferences to be made about the forces that act on nascent cancer clones as they evolve and have shown insight into the mutational processes that generate genetic variation. Here we review our emerging understanding of the dynamic evolution of the cancer genome and of the implications for basic cancer biology and the development of antitumour therapy.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
