

The French Gaucher's disease registry: clinical characteristics, complications and treatment of 562 patients
- PMID: 23046562
- PMCID: PMC3526516
- DOI: 10.1186/1750-1172-7-77
The French Gaucher's disease registry: clinical characteristics, complications and treatment of 562 patients
Abstract
Background: Clinical features, complications and treatments of Gaucher's disease (GD), a rare autosomal-recessive disorder due to a confirmed lysosomal enzyme (glucocerebrosidase) deficiency, are described.
Methods: All patients with known GD, living in France, with ≥ 1 consultations (1980-2010), were included in the French GD registry, yielding the following 4 groups: the entire cohort, with clinical description; and its subgroups: patients with ≥ 1 follow-up visits, to investigate complications; recently followed (2009-2010) patients; and patients treated during 2009-2010, to examine complications before and during treatment. Data are expressed as medians (range) for continuous variables and numbers (%) for categorical variables.
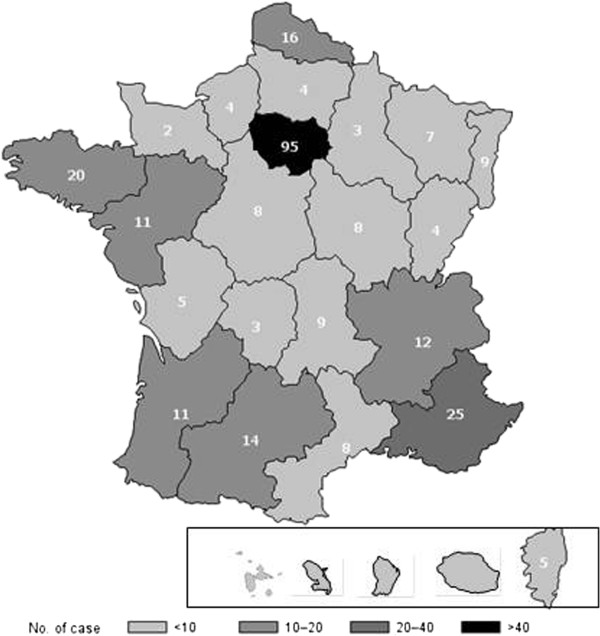
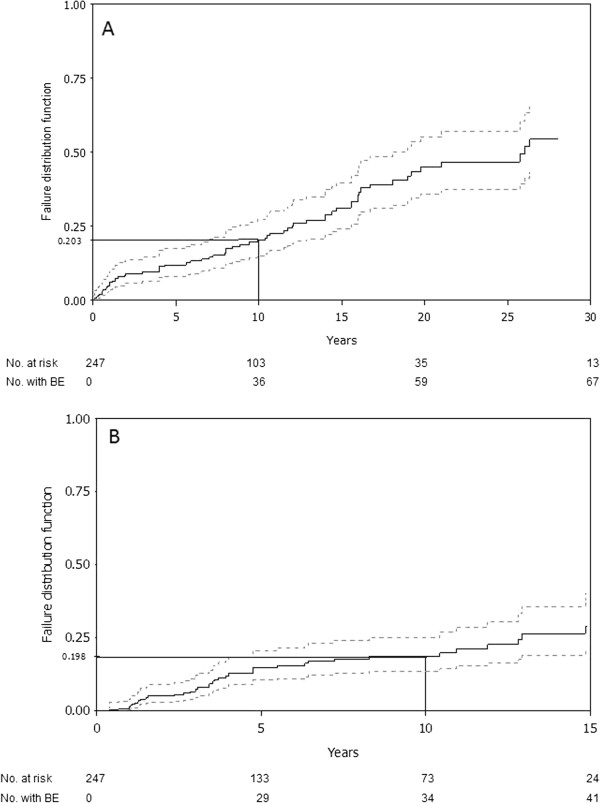
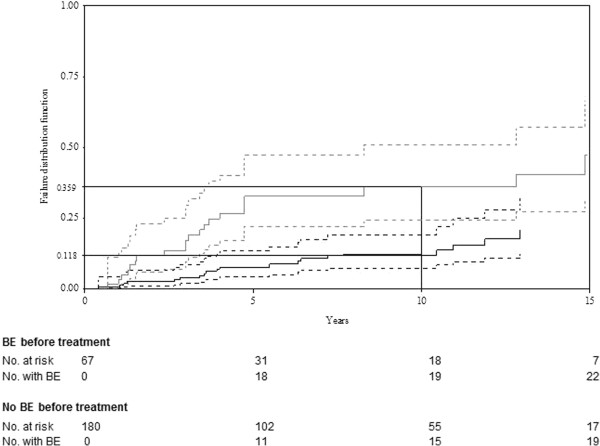
Results: Among the 562 registry patients, 265 (49.6%) were females; 454 (85.0%) had type 1, 22 (4.1%) type 2, 37 (6.9%) perinatal-lethal type and 21 (3.9%) type 3. Median ages at first GD symptoms and diagnosis, respectively, were 15 (0-77) and 22 (0-84) years for all types. The first symptom diagnosing GD was splenomegaly and/or thrombocytopenia (37.6% and 26.3%, respectively). Bone-marrow aspiration and/or biopsy yielded the diagnosis for 54.7% of the patients, with enzyme deficiency confirming GD for all patients. Birth incidence rate was estimated at 1/50,000 and prevalence at 1/136,000. For the 378 followed patients, median follow-up was 16.2 (0.1-67.6) years. Major clinical complications were bone events (BE; avascular necrosis, bone infarct or pathological fracture) for 109 patients, splenectomy for 104, and Parkinson's disease for 14; 38 patients died (neurological complications for 15 type-2 and 3 type-3 patients, GD complications for 11 type-1 and another disease for 9 type-1 patients). Forty-six had monoclonal gammopathy. Among 283 recently followed patients, 36 were untreated and 247 had been treated during 2009-2010; 216 patients received treatment in December 2010 (126 with imiglucerase, 45 velaglucerase, 24 taliglucerase, 21 miglustat). BE occurred before (130 in 67 patients) and under treatment (60 in 41 patients) with respective estimated frequencies (95% CI) of first BE at 10 years of 20.3% (14.1%-26.5%) and 19.8% (13.5%-26.1%).
Conclusion: This registry enabled the epidemiological description of GD in France and showed that BE occur even during treatment.
Figures
References
-
- Christomanou H, Aignesberger A, Linke RP. Immunochemical characterization of two activator proteins stimulating enzymic sphingomyelin degradation in vitro. Absence of one of them in a human Gaucher disease variant. Biol Chem Hoppe Seyler. 1986;367:879–890. doi: 10.1515/bchm3.1986.367.2.879. - DOI - PubMed
-
- Qi X, Grabowski GA. Molecular and cell biology of acid beta-glucosidase and prosaposin. Prog Nucleic Acid Res Mol Biol. 2001;66:203–239. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
