

The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations
- PMID: 23049765
- PMCID: PMC3458854
- DOI: 10.1371/journal.pone.0045001
The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations
Abstract
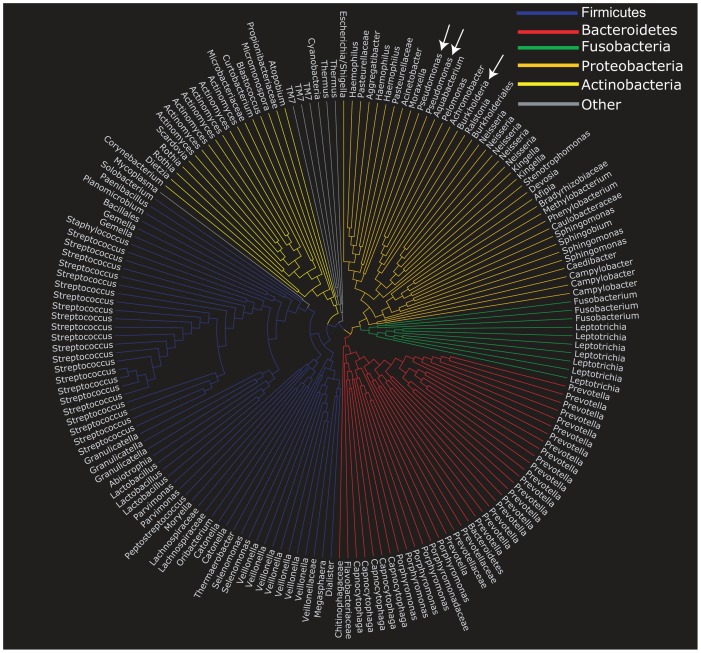
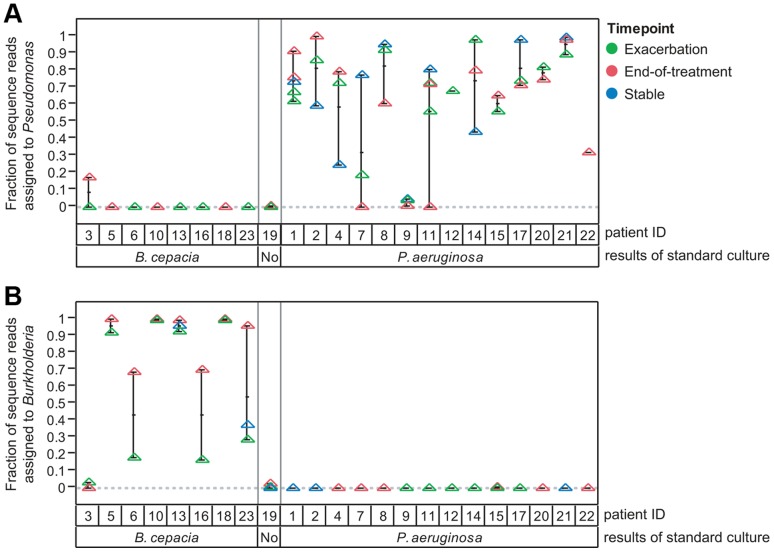
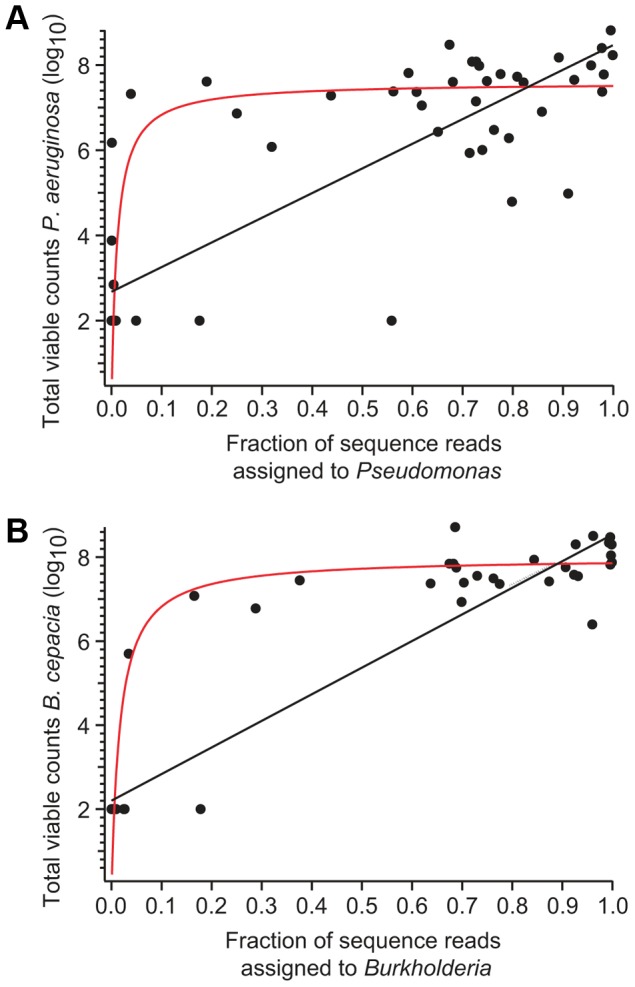
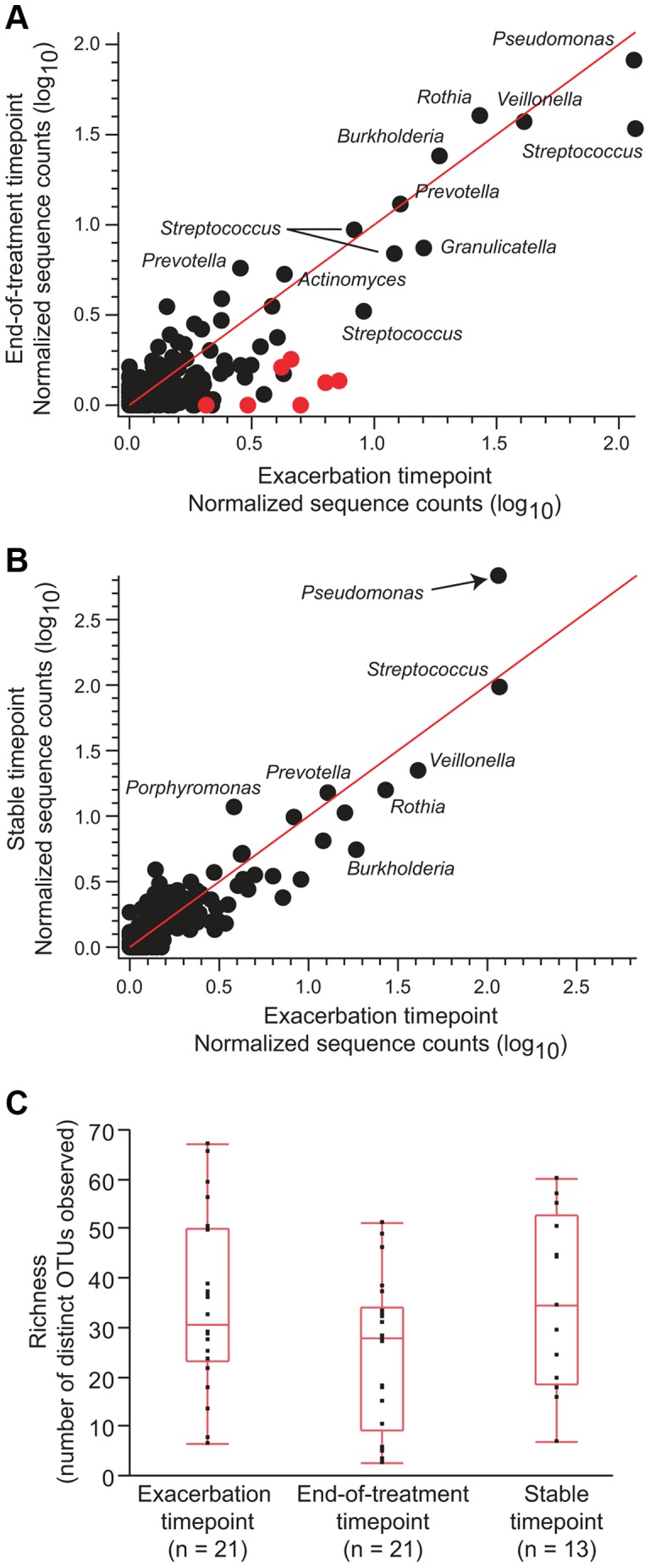
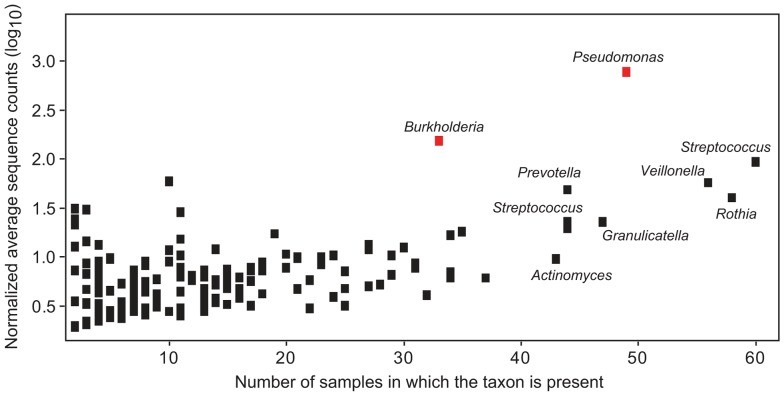
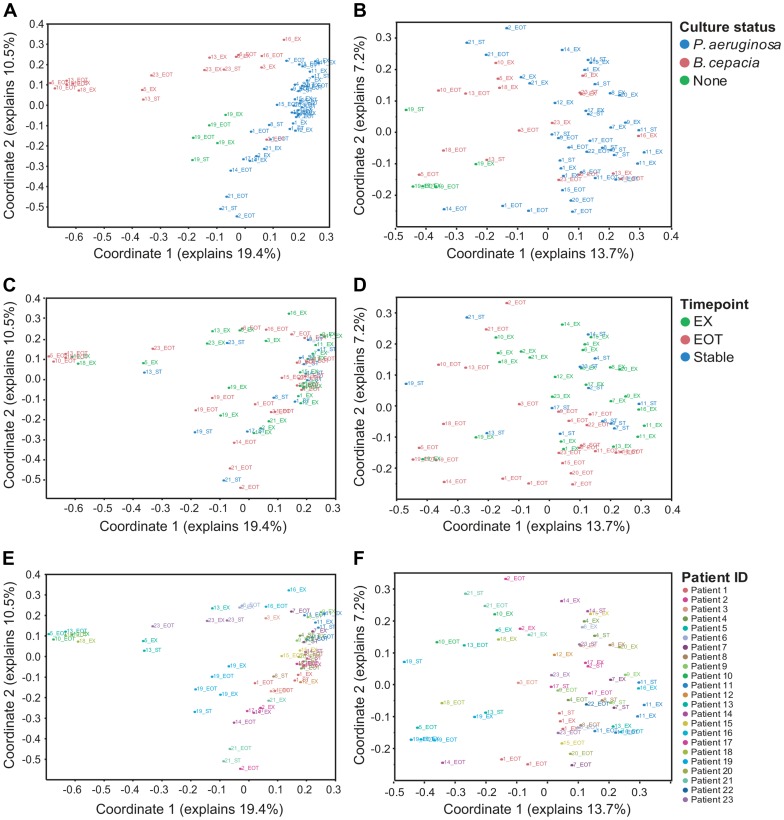
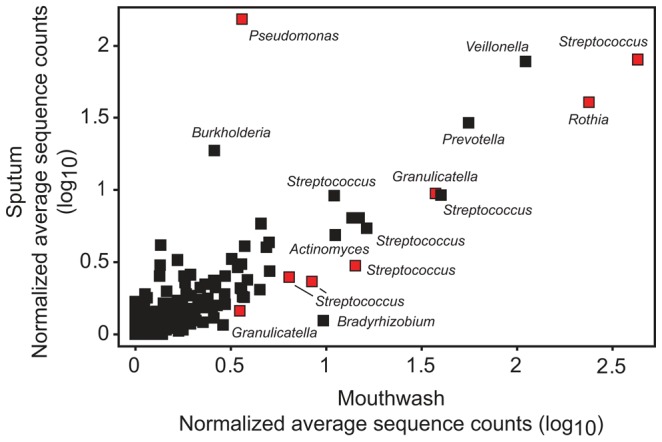
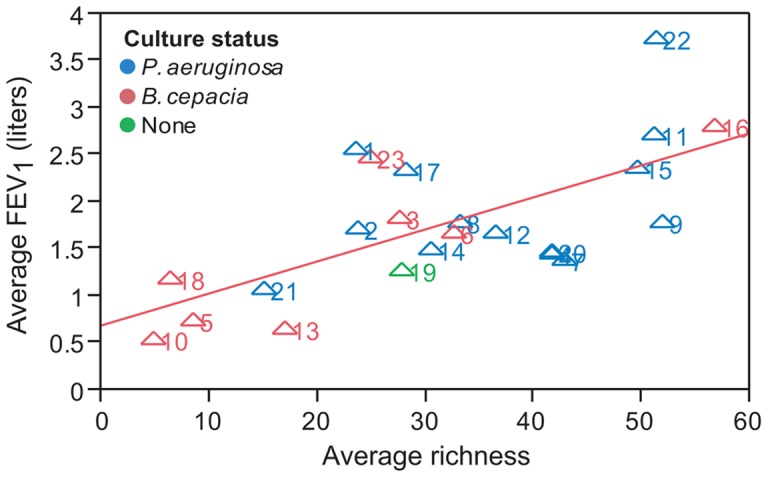
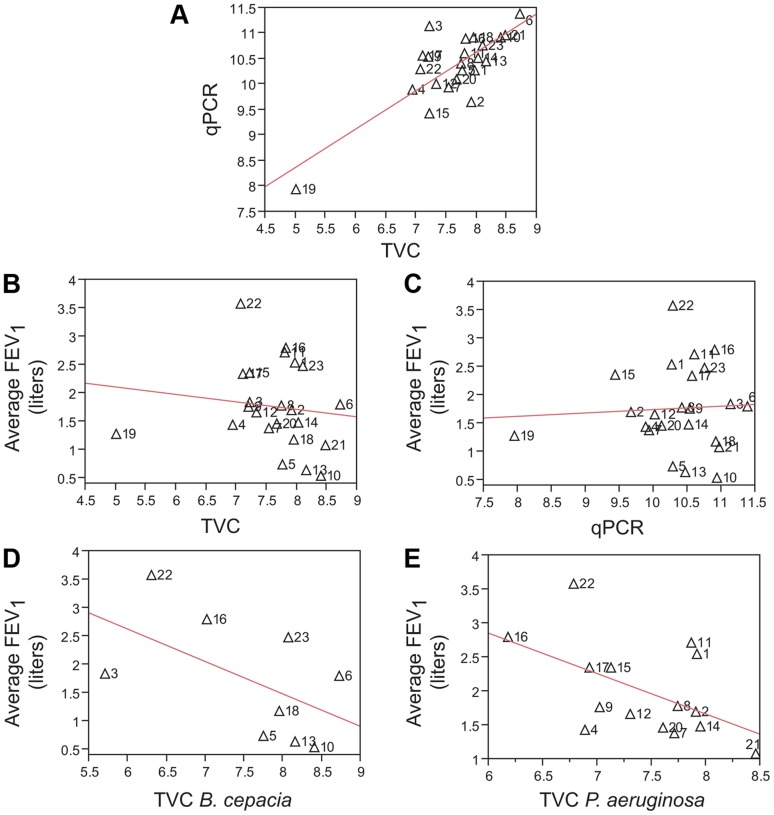
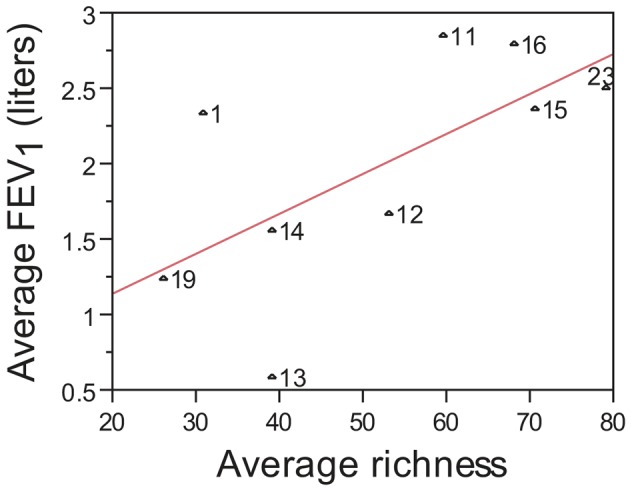
Cystic fibrosis (CF) is characterized by defective mucociliary clearance and chronic airway infection by a complex microbiota. Infection, persistent inflammation and periodic episodes of acute pulmonary exacerbation contribute to an irreversible decline in CF lung function. While the factors leading to acute exacerbations are poorly understood, antibiotic treatment can temporarily resolve pulmonary symptoms and partially restore lung function. Previous studies indicated that exacerbations may be associated with changes in microbial densities and the acquisition of new microbial species. Given the complexity of the CF microbiota, we applied massively parallel pyrosequencing to identify changes in airway microbial community structure in 23 adult CF patients during acute pulmonary exacerbation, after antibiotic treatment and during periods of stable disease. Over 350,000 sequences were generated, representing nearly 170 distinct microbial taxa. Approximately 60% of sequences obtained were from the recognized CF pathogens Pseudomonas and Burkholderia, which were detected in largely non-overlapping patient subsets. In contrast, other taxa including Prevotella, Streptococcus, Rothia and Veillonella were abundant in nearly all patient samples. Although antibiotic treatment was associated with a small decrease in species richness, there was minimal change in overall microbial community structure. Furthermore, microbial community composition was highly similar in patients during an exacerbation and when clinically stable, suggesting that exacerbations may represent intrapulmonary spread of infection rather than a change in microbial community composition. Mouthwash samples, obtained from a subset of patients, showed a nearly identical distribution of taxa as expectorated sputum, indicating that aspiration may contribute to colonization of the lower airways. Finally, we observed a strong correlation between low species richness and poor lung function. Taken together, these results indicate that the adult CF lung microbiome is largely stable through periods of exacerbation and antibiotic treatment and that short-term compositional changes in the airway microbiota do not account for CF pulmonary exacerbations.
Conflict of interest statement
Figures
Similar articles
-
Inflammation and airway microbiota during cystic fibrosis pulmonary exacerbations.PLoS One. 2013 Apr 30;8(4):e62917. doi: 10.1371/journal.pone.0062917. Print 2013. PLoS One. 2013. PMID: 23646159 Free PMC article.
-
Measures of Cystic Fibrosis Airway Microbiota during Periods of Clinical Stability.Ann Am Thorac Soc. 2019 Dec;16(12):1534-1542. doi: 10.1513/AnnalsATS.201903-270OC. Ann Am Thorac Soc. 2019. PMID: 31415187 Free PMC article.
-
Analysis of changes in diversity and abundance of the microbial community in a cystic fibrosis patient over a multiyear period.J Clin Microbiol. 2015 Jan;53(1):237-47. doi: 10.1128/JCM.02555-14. Epub 2014 Nov 12. J Clin Microbiol. 2015. PMID: 25392361 Free PMC article.
-
Antibiotic treatment of multidrug-resistant organisms in cystic fibrosis.Am J Respir Med. 2003;2(4):321-32. doi: 10.1007/BF03256660. Am J Respir Med. 2003. PMID: 14719998 Review.
-
Antimicrobial resistance in the respiratory microbiota of people with cystic fibrosis.Lancet. 2014 Aug 23;384(9944):703-13. doi: 10.1016/S0140-6736(14)61137-5. Lancet. 2014. PMID: 25152272 Review.
Cited by
-
Data Mining of Lung Microbiota in Cystic Fibrosis Patients.PLoS One. 2016 Oct 14;11(10):e0164510. doi: 10.1371/journal.pone.0164510. eCollection 2016. PLoS One. 2016. PMID: 27741283 Free PMC article.
-
Impact of multidrug-resistant organisms on patients considered for lung transplantation.Infect Dis Clin North Am. 2013 Jun;27(2):343-58. doi: 10.1016/j.idc.2013.02.006. Epub 2013 Apr 17. Infect Dis Clin North Am. 2013. PMID: 23714344 Free PMC article. Review.
-
Antimicrobial therapies for prevention of recurrent acute exacerbations of COPD (AECOPD): beyond the guidelines.Respir Res. 2022 Mar 14;23(1):58. doi: 10.1186/s12931-022-01947-5. Respir Res. 2022. PMID: 35287677 Free PMC article. Review.
-
Cystic fibrosis respiratory microbiota: unraveling complexity to inform clinical practice.Expert Rev Respir Med. 2018 Oct;12(10):857-865. doi: 10.1080/17476348.2018.1513331. Epub 2018 Sep 3. Expert Rev Respir Med. 2018. PMID: 30118374 Free PMC article. Review.
-
Antibiotics Drive Expansion of Rare Pathogens in a Chronic Infection Microbiome Model.mSphere. 2022 Oct 26;7(5):e0031822. doi: 10.1128/msphere.00318-22. Epub 2022 Aug 16. mSphere. 2022. PMID: 35972133 Free PMC article.
References
-
- Boyle MP (2007) Adult cystic fibrosis. JAMA 298: 1787–1793. - PubMed
-
- Dodge JA, Lewis PA, Stanton M, Wilsher J (2007) Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J 29: 522–526. - PubMed
-
- Hodson ME, Simmonds NJ, Warwick WJ, Tullis E, Castellani C, et al. (2008) An international/multicentre report on patients with cystic fibrosis (CF) over the age of 40 years. J Cyst Fibros 7: 537–542. - PubMed
-
- Razvi S, Quittell L, Sewall A, Quinton H, Marshall B, et al. (2009) Respiratory microbiology of patients with cystic fibrosis in the United States, 1995 to 2005. Chest 136: 1554–1560. - PubMed
