

Comparative genomics of the classical Bordetella subspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens
- PMID: 23051057
- PMCID: PMC3533505
- DOI: 10.1186/1471-2164-13-545
Comparative genomics of the classical Bordetella subspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens
Abstract
Background: The classical Bordetella subspecies are phylogenetically closely related, yet differ in some of the most interesting and important characteristics of pathogens, such as host range, virulence and persistence. The compelling picture from previous comparisons of the three sequenced genomes was of genome degradation, with substantial loss of genome content (up to 24%) associated with adaptation to humans.
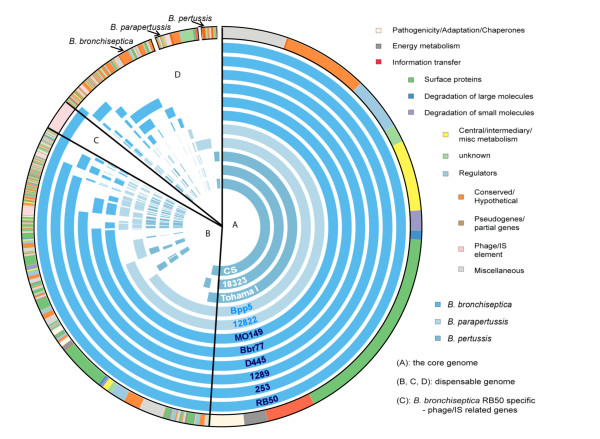
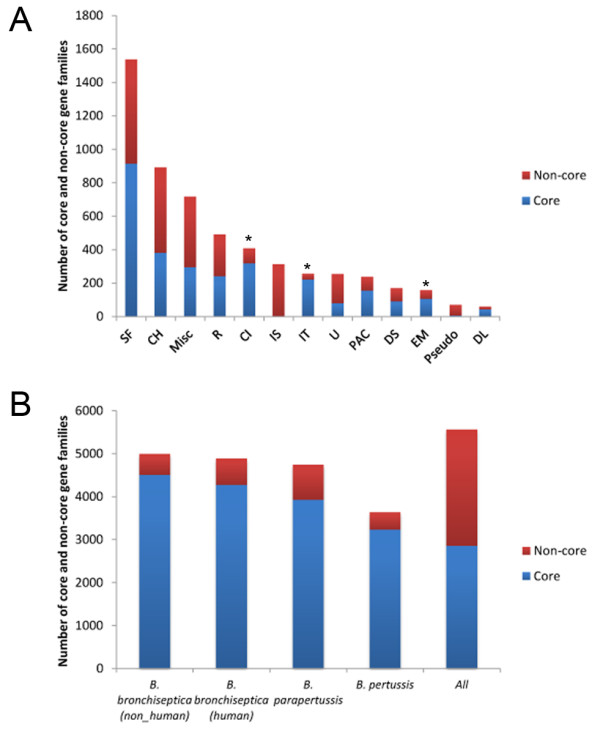
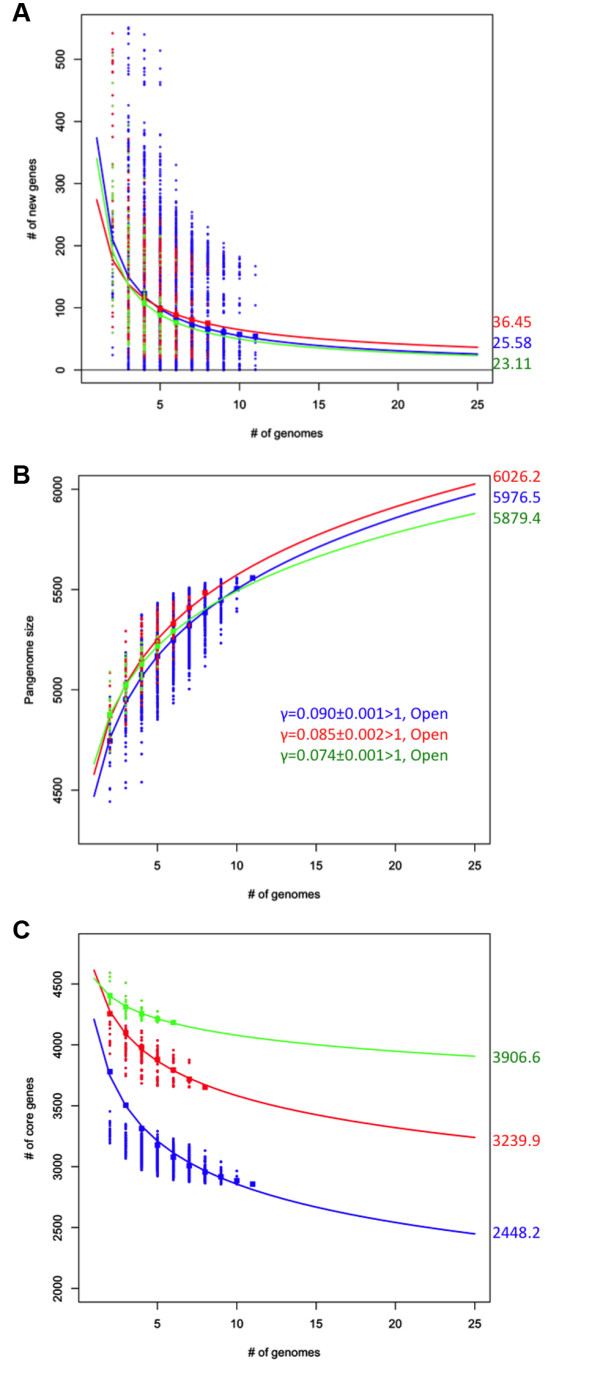
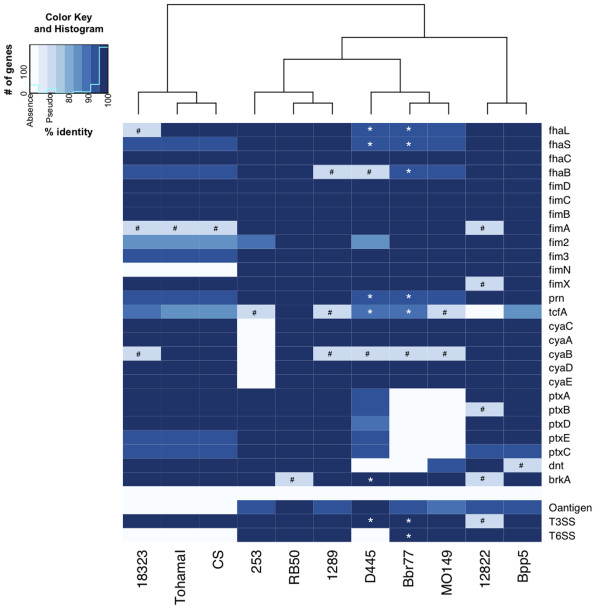
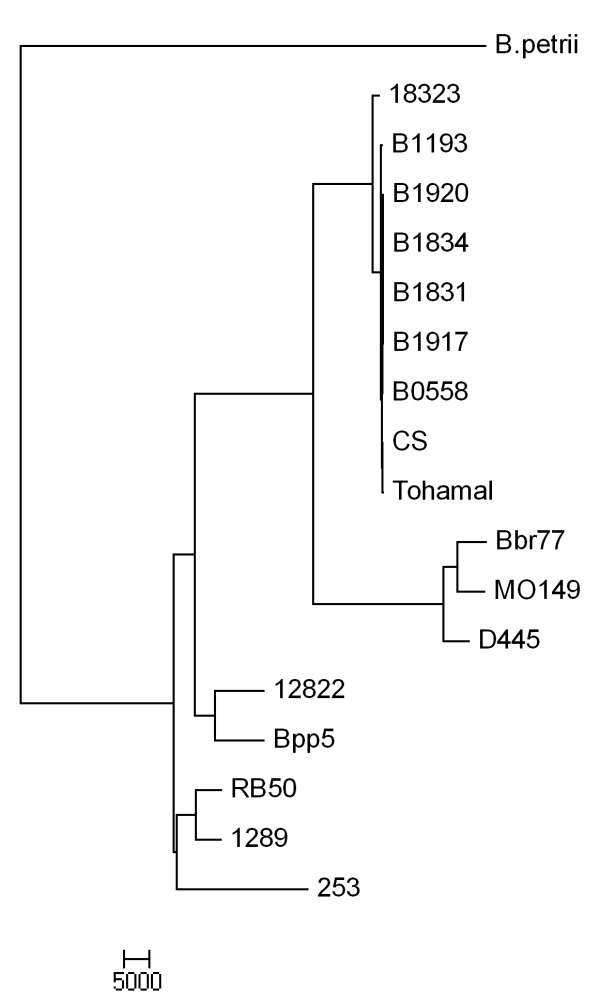
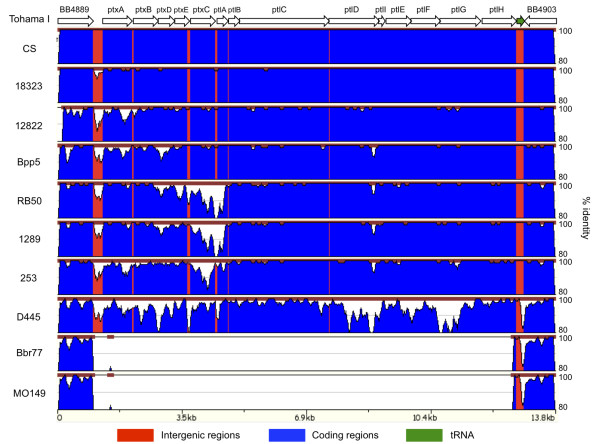
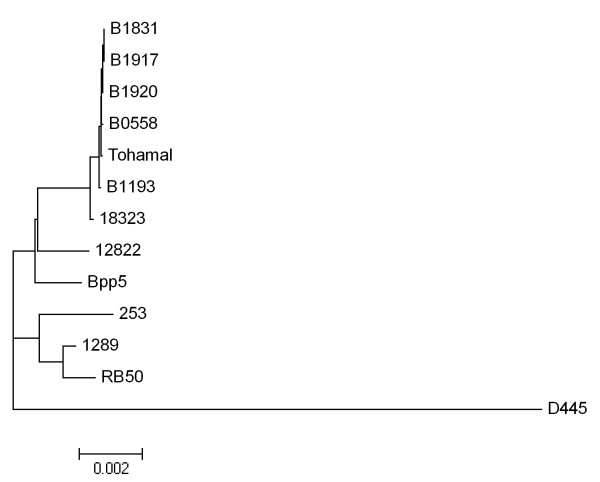
Results: For a more comprehensive picture of lineage evolution, we employed comparative genomic and phylogenomic analyses using seven additional diverse, newly sequenced Bordetella isolates. Genome-wide single nucleotide polymorphism (SNP) analysis supports a reevaluation of the phylogenetic relationships between the classical Bordetella subspecies, and suggests a closer link between ovine and human B. parapertussis lineages than has been previously proposed. Comparative analyses of genome content revealed that only 50% of the pan-genome is conserved in all strains, reflecting substantial diversity of genome content in these closely related pathogens that may relate to their different host ranges, virulence and persistence characteristics. Strikingly, these analyses suggest possible horizontal gene transfer (HGT) events in multiple loci encoding virulence factors, including O-antigen and pertussis toxin (Ptx). Segments of the pertussis toxin locus (ptx) and its secretion system locus (ptl) appear to have been acquired by the classical Bordetella subspecies and are divergent in different lineages, suggesting functional divergence in the classical Bordetellae.
Conclusions: Together, these observations, especially in key virulence factors, reveal that multiple mechanisms, such as point mutations, gain or loss of genes, as well as HGTs, contribute to the substantial phenotypic diversity of these versatile subspecies in various hosts.
Figures
References
-
- van der Zee A, Groenendijk H, Peeters M, Mooi FR. The differentiation of Bordetella parapertussis and Bordetella bronchiseptica from humans and animals as determined by DNA polymorphism mediated by two different insertion sequence elements suggests their phylogenetic relationship. Int J Syst Bacteriol. 1996;46:640–647. doi: 10.1099/00207713-46-3-640. - DOI - PubMed
-
- Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MTG, Churcher CM, Bentley SD, Mungall KL, Cerdeño-Tárraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O’Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis, and Bordetella bronchiseptica. Nat Genet. 2003;35:32–40. doi: 10.1038/ng1227. - DOI - PubMed
-
- Sebaihia M, Preston A, Maskell DJ, Kuzmiak H, Connell TD, King ND, Orndorff PE, Miyamoto DM, Thomson NR, Harris D, Goble A, Lord A, Murphy L, Quail MA, Rutter S, Squares R, Squares S, Woodward J, Parkhill J, Temple LM. Comparison of the genome sequence of the poultry pathogen Bordetella avium with those of B. bronchiseptica, B. pertussis, and B. parapertussis reveals extensive diversity in surface structures associated with host interaction. J Bacteriol. 2006;188:6002–6015. doi: 10.1128/JB.01927-05. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
