

Mechanisms of telomere loss and their consequences for chromosome instability
- PMID: 23061048
- PMCID: PMC3463808
- DOI: 10.3389/fonc.2012.00135
Mechanisms of telomere loss and their consequences for chromosome instability
Abstract
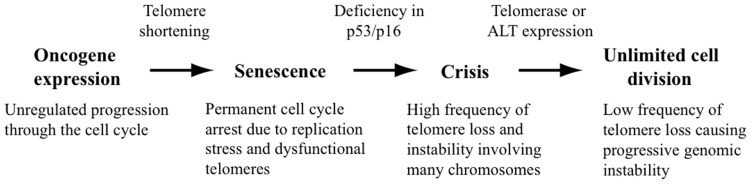
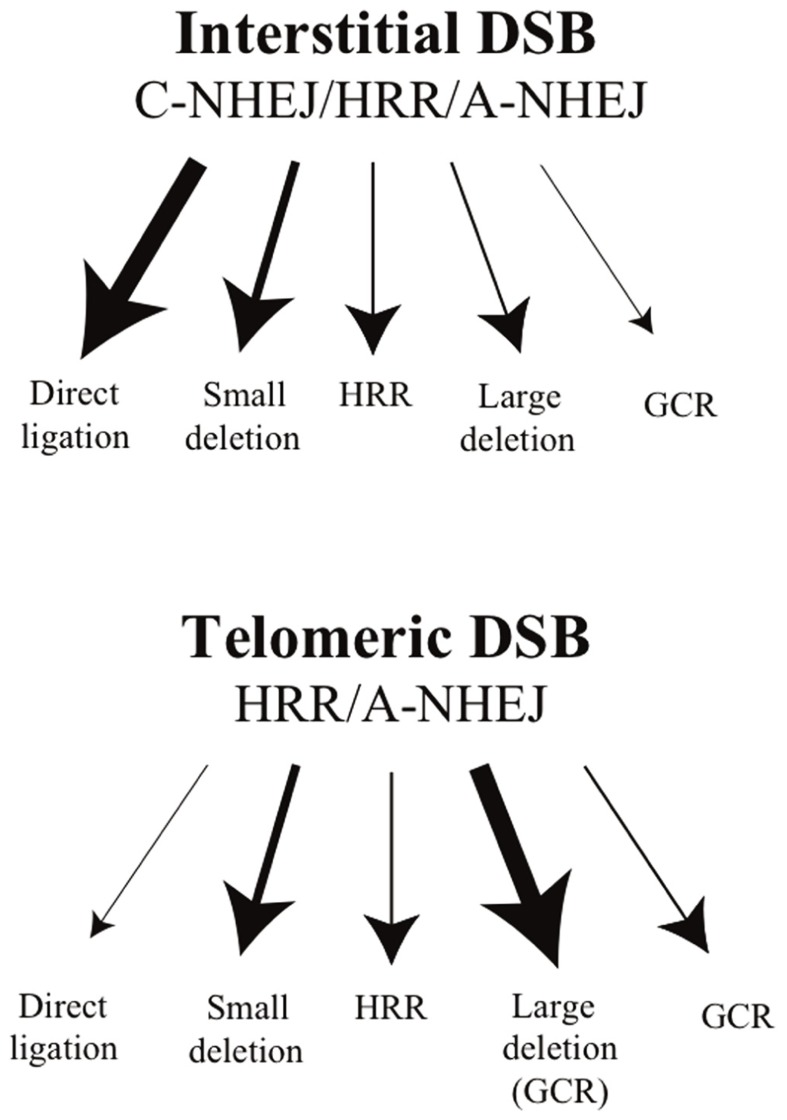
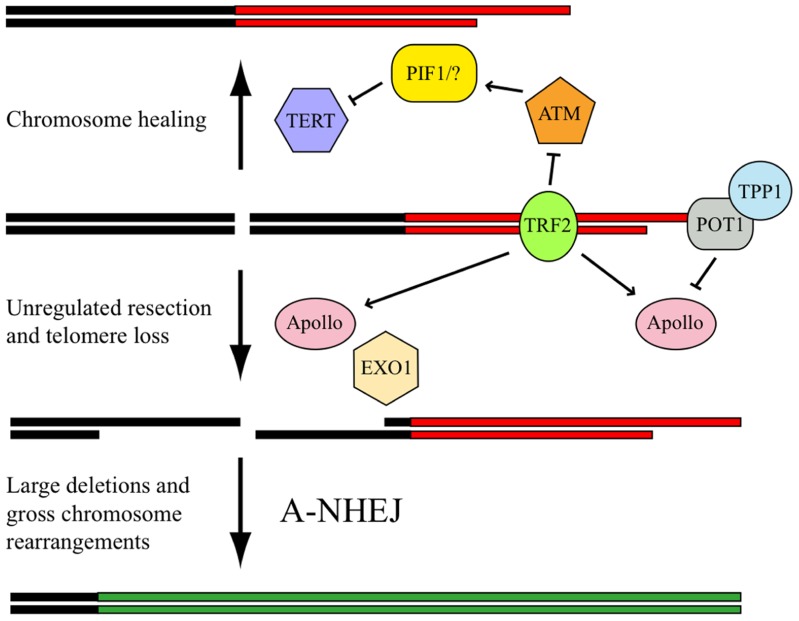
The ends of chromosomes in mammals, called telomeres, are composed of a 6-bp repeat sequence, TTAGGG, which is added on by the enzyme telomerase. In combination with a protein complex called shelterin, these telomeric repeat sequences form a cap that protects the ends of chromosomes. Due to insufficient telomerase expression, telomeres shorten gradually with each cell division in human somatic cells, which limits the number of times they can divide. The extensive cell division involved in cancer cell progression therefore requires that cancer cells must acquire the ability to maintain telomeres, either through expression of telomerase, or through an alternative mechanism involving recombination. It is commonly thought that the source of many chromosome rearrangements in cancer cells is a result of the extensive telomere shortening that occurs prior to the expression of telomerase. However, despite the expression of telomerase, tumor cells can continue to show chromosome instability due to telomere loss. Dysfunctional telomeres in cancer cells can result from oncogene-induced replication stress, which results in double-strand breaks (DSBs) at fragile sites, including telomeres. DSBs near telomeres are especially prone to chromosome rearrangements, because telomeric regions are deficient in DSB repair. The deficiency in DSB repair near telomeres is also an important mechanism for ionizing radiation-induced replicative senescence in normal human cells. In addition, DSBs near telomeres can result in chromosome instability in mouse embryonic stem cells, suggesting that telomere loss can contribute to heritable chromosome rearrangements. Consistent with this possibility, telomeric regions in humans are highly heterogeneous, and chromosome rearrangements near telomeres are commonly involved in human genetic disease. Understanding the mechanisms of telomere loss will therefore provide important insights into both human cancer and genetic disease.
Keywords: chromosome instability; double-strand break repair; gross chromosomal rearrangement; sister chromatid fusion; telomere.
Figures
References
-
- Allen C., Halbrook J., Nickoloff J. A. (2003). Interactive competition between homologous recombination and non-homologous end joining. Mol. Cancer Res. 1 913–920 - PubMed
-
- Amiard S., Doudeau M., Pinte S., Poulet A., Lenain C., Faivre-Moskalenko C., et al. (2007). A topological mechanism for TRF2-enhanced strand invasion. Nat. Struct. Mol. Biol. 14 147–154 - PubMed
-
- Anderson B. H., Kasher P. R., Mayer J., Szynkiewicz M., Jenkinson E. M., Bhaskar S. S., et al. (2012). Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat. Genet. 44 338–342 - PubMed
-
- Artandi S. E., Chang S., Lee S.-L., Alson S., Gottlieb G. J., Chin L., et al. (2000). Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406 641–645 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous