

SeqFold: genome-scale reconstruction of RNA secondary structure integrating high-throughput sequencing data
- PMID: 23064747
- PMCID: PMC3561878
- DOI: 10.1101/gr.138545.112
SeqFold: genome-scale reconstruction of RNA secondary structure integrating high-throughput sequencing data
Abstract
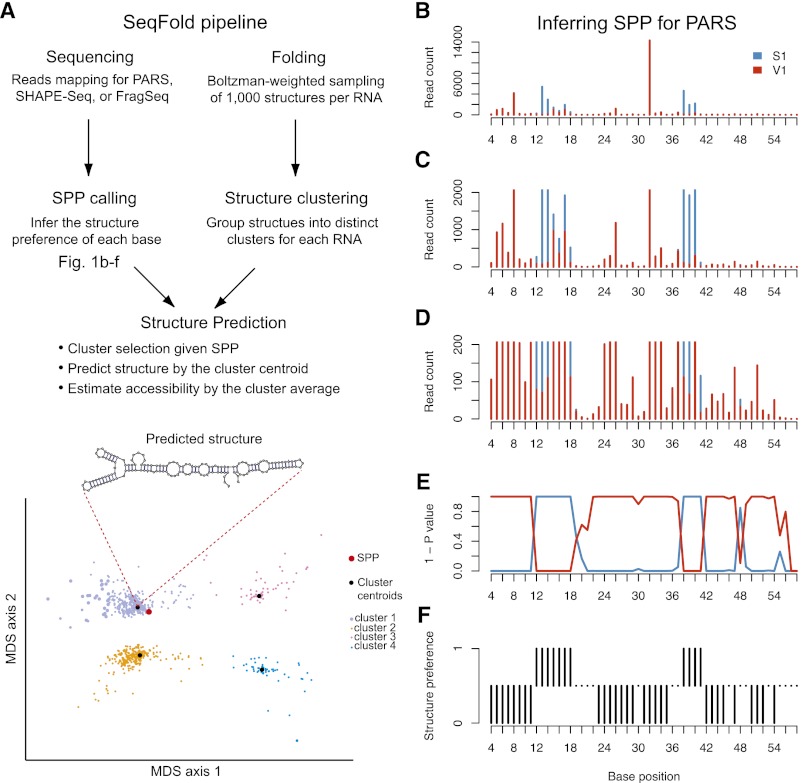
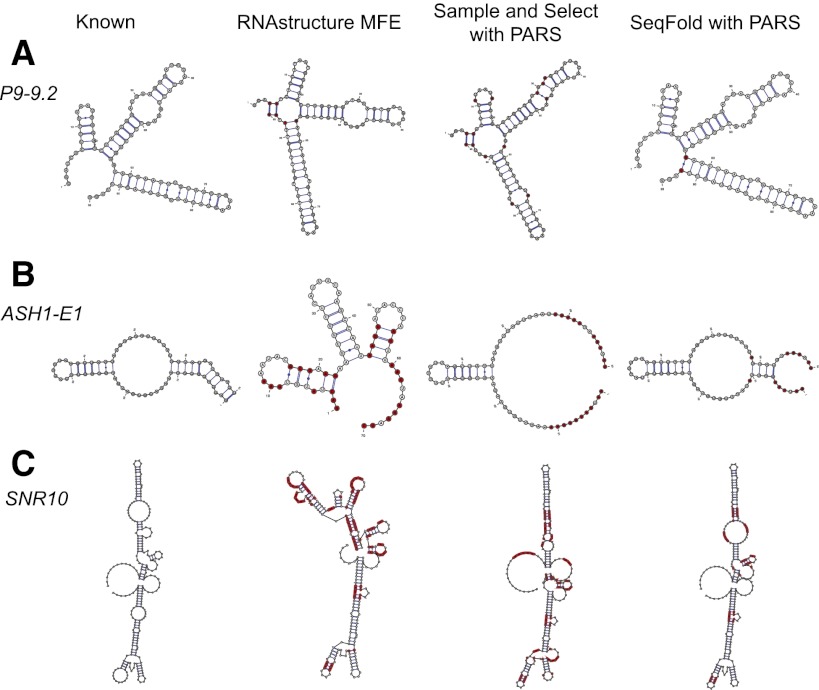
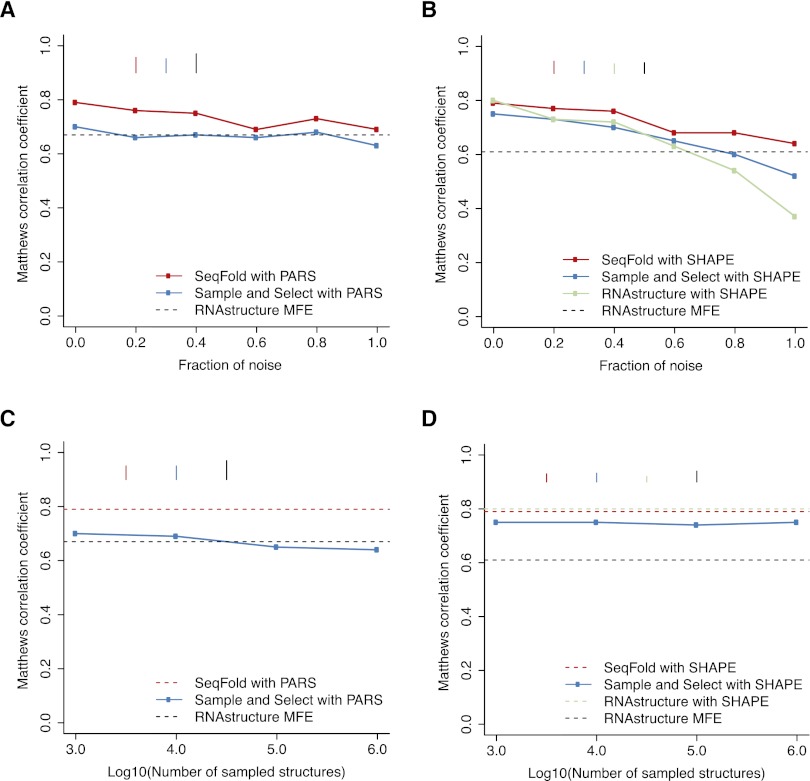
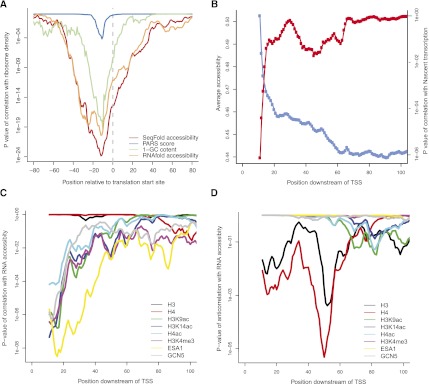
We present an integrative approach, SeqFold, that combines high-throughput RNA structure profiling data with computational prediction for genome-scale reconstruction of RNA secondary structures. SeqFold transforms experimental RNA structure information into a structure preference profile (SPP) and uses it to select stable RNA structure candidates representing the structure ensemble. Under a high-dimensional classification framework, SeqFold efficiently matches a given SPP to the most likely cluster of structures sampled from the Boltzmann-weighted ensemble. SeqFold is able to incorporate diverse types of RNA structure profiling data, including parallel analysis of RNA structure (PARS), selective 2'-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq), fragmentation sequencing (FragSeq) data generated by deep sequencing, and conventional SHAPE data. Using the known structures of a wide range of mRNAs and noncoding RNAs as benchmarks, we demonstrate that SeqFold outperforms or matches existing approaches in accuracy and is more robust to noise in experimental data. Application of SeqFold to reconstruct the secondary structures of the yeast transcriptome reveals the diverse impact of RNA secondary structure on gene regulation, including translation efficiency, transcription initiation, and protein-RNA interactions. SeqFold can be easily adapted to incorporate any new types of high-throughput RNA structure profiling data and is widely applicable to analyze RNA structures in any transcriptome.
Figures
References
-
- Alkemar G, Nygard O 2006. Probing the secondary structure of expansion segment ES6 in 18S ribosomal RNA. Biochemistry 45: 8067–8078 - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ 1990. Basic local alignment search tool. J Mol Biol 215: 403–410 - PubMed
-
- Aviv T, Lin Z, Ben-Ari G, Smibert CA, Sicheri F 2006. Sequence-specific recognition of RNA hairpins by the SAM domain of Vts1p. Nat Struct Mol Biol 13: 168–176 - PubMed
-
- Baldi P, Brunak S, Chauvin Y, Andersen C, Nielsen H 2000. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 16: 412–424 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous