

Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy
- PMID: 23065789
- PMCID: PMC3470714
- DOI: 10.1093/brain/aws240
Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy
Abstract
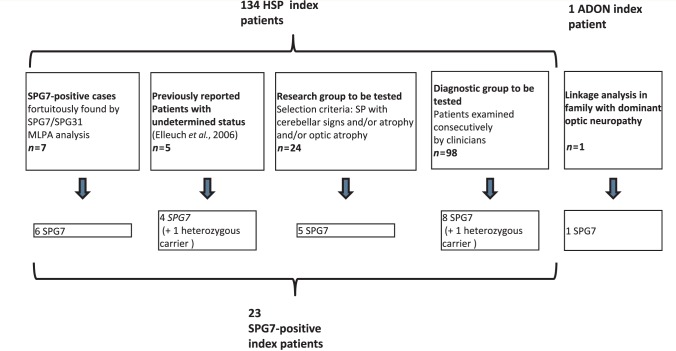
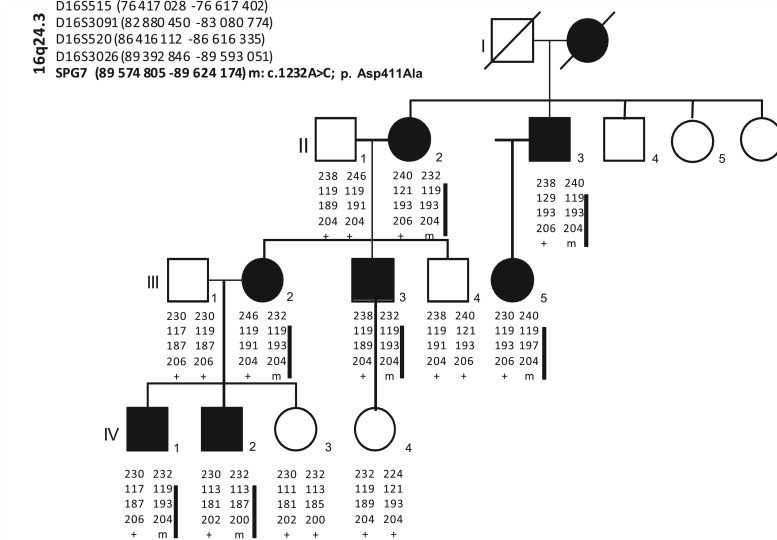
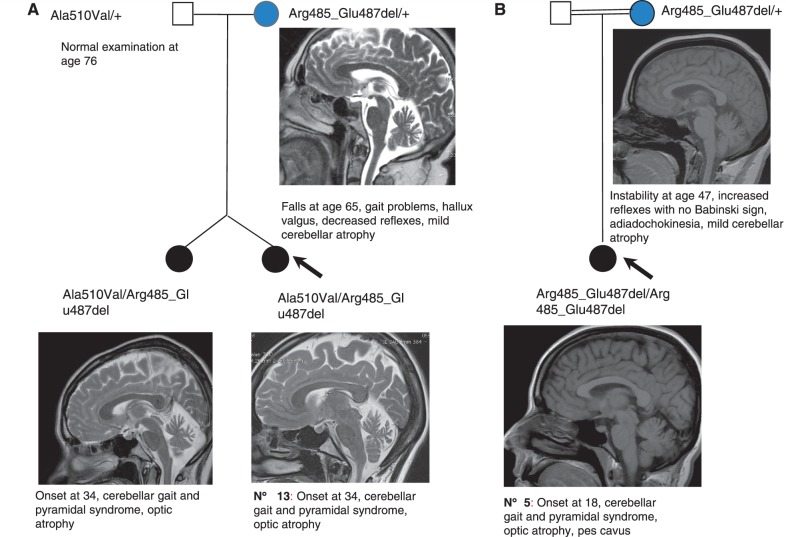
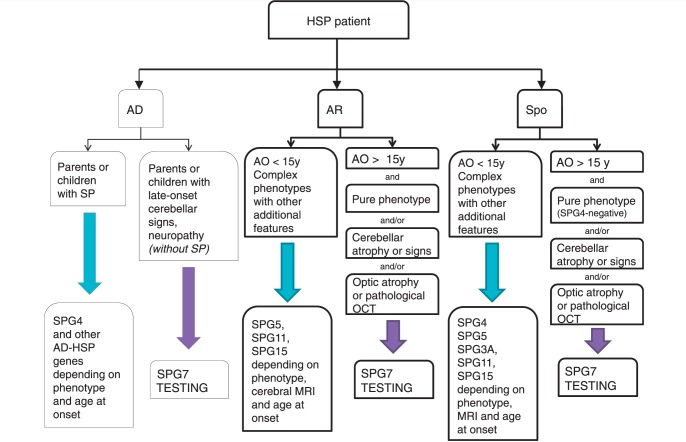
Mutations in the spastic paraplegia 7 (SPG7) gene encoding paraplegin are responsible for autosomal recessive hereditary spasticity. We screened 135 unrelated index cases, selected in five different settings: SPG7-positive patients detected during SPG31 analysis using SPG31/SPG7 multiplex ligation-dependent probe amplification (n = 7); previously reported ambiguous SPG7 cases (n = 5); patients carefully selected on the basis of their phenotype (spasticity of the lower limbs with cerebellar signs and/or cerebellar atrophy on magnetic resonance imaging/computer tomography scan and/or optic neuropathy and without other signs) (n = 24); patients with hereditary spastic paraparesis referred consecutively from attending neurologists and the national reference centre in a diagnostic setting (n = 98); and the index case of a four-generation family with autosomal dominant optic neuropathy but no spasticity linked to the SPG7 locus. We identified two SPG7 mutations in 23/134 spastic patients, 21% of the patients selected according to phenotype but only 8% of those referred directly. Our results confirm the pathogenicity of Ala510Val, which was the most frequent mutation in our series (65%) and segregated at the homozygous state with spastic paraparesis in a large family with autosomal recessive inheritance. All SPG7-positive patients tested had optic neuropathy or abnormalities revealed by optical coherence tomography, indicating that abnormalities in optical coherence tomography could be a clinical biomarker for SPG7 testing. In addition, the presence of late-onset very slowly progressive spastic gait (median age 39 years, range 18-52 years) associated with cerebellar ataxia (39%) or cerebellar atrophy (47%) constitute, with abnormal optical coherence tomography, key features pointing towards SPG7-testing. Interestingly, three relatives of patients with heterozygote SPG7 mutations had cerebellar signs and atrophy, or peripheral neuropathy, but no spasticity of the lower limbs, suggesting that SPG7 mutations at the heterozygous state might predispose to late-onset neurodegenerative disorders, mimicking autosomal dominant inheritance. Finally, a novel missense SPG7 mutation at the heterozygous state (Asp411Ala) was identified as the cause of autosomal dominant optic neuropathy in a large family, indicating that some SPG7 mutations can occasionally be dominantly inherited and be an uncommon cause of isolated optic neuropathy. Altogether, these results emphasize the clinical variability associated with SPG7 mutations, ranging from optic neuropathy to spastic paraplegia, and support the view that SPG7 screening should be carried out in both conditions.
Figures
References
-
- Arnoldi A, Tonelli A, Crippa F, Villani G, Pacelli C, Sironi M, et al. A clinical, genetic, and biochemical characterization of SPG7 mutations in a large cohort of patients with hereditary spastic paraplegia. Hum Mutat. 2008;29:522–31. - PubMed
-
- Banfi S, Bassi MT, Andolfi G, Marchitiello A, Zanotta S, Ballabio A, et al. Identification and characterization of AFG3L2, a novel paraplegin-related gene. Genomics. 1999;59:51–8. - PubMed
