

Critical role of neuronal pentraxin 1 in mitochondria-mediated hypoxic-ischemic neuronal injury
- PMID: 23069675
- PMCID: PMC3534917
- DOI: 10.1016/j.nbd.2012.10.003
Critical role of neuronal pentraxin 1 in mitochondria-mediated hypoxic-ischemic neuronal injury
Abstract
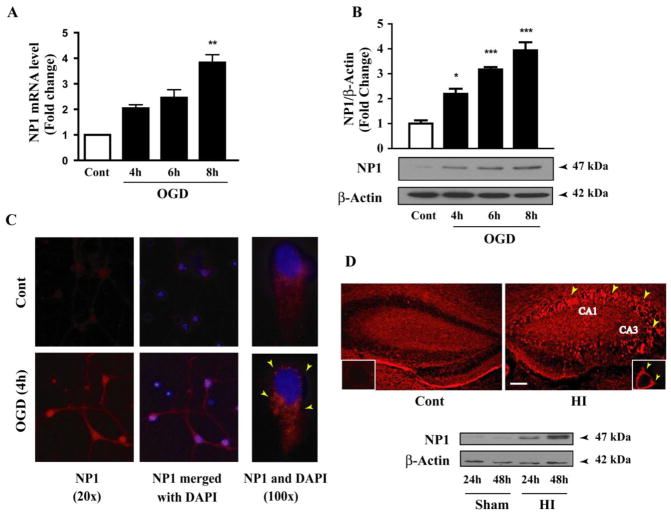
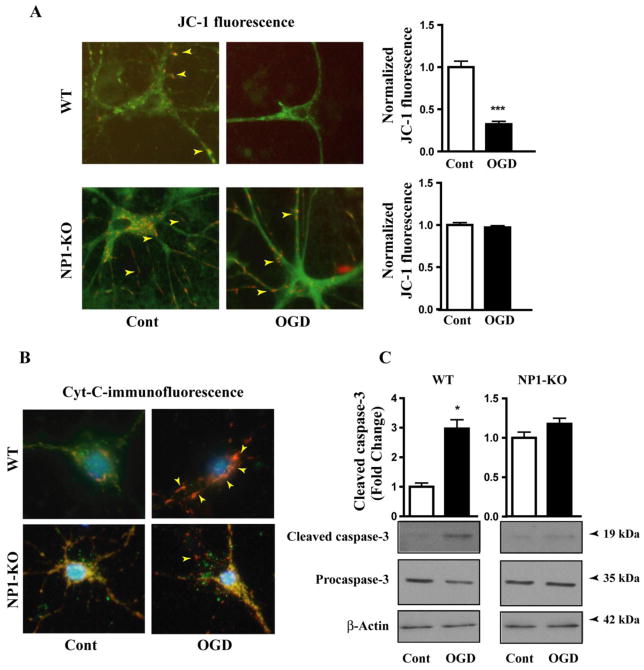
Developing brain is highly susceptible to hypoxic-ischemic (HI) injury leading to severe neurological disabilities in surviving infants and children. Previously, we have reported induction of neuronal pentraxin 1 (NP1), a novel neuronal protein of long-pentraxin family, following HI neuronal injury. Here, we investigated how this specific signal is propagated to cause the HI neuronal death. We used wild-type (WT) and NP1 knockout (NP1-KO) mouse hippocampal cultures, modeled in vitro following exposure to oxygen glucose deprivation (OGD), and in vivo neonatal (P9-10) mouse model of HI brain injury. Our results show induction of NP1 in primary hippocampal neurons following OGD exposure (4-8 h) and in the ipsilateral hippocampal CA1 and CA3 regions at 24-48 h post-HI compared to the contralateral side. We also found increased PTEN activity concurrent with OGD time-dependent (4-8 h) dephosphorylation of Akt (Ser473) and GSK-3β (Ser9). OGD also caused a time-dependent decrease in the phosphorylation of Bad (Ser136), and Bax protein levels. Immunofluorescence staining and subcellular fractionation analyses revealed increased mitochondrial translocation of Bad and Bax proteins from cytoplasm following OGD (4 h) and simultaneously increased release of Cyt C from mitochondria followed by activation of caspase-3. NP1 protein was immunoprecipitated with Bad and Bax proteins; OGD caused increased interactions of NP1 with Bad and Bax, thereby, facilitating their mitochondrial translocation and dissipation of mitochondrial membrane potential (ΔΨ(m)). This NP1 induction preceded the increased mitochondrial release of cytochrome C (Cyt C) into the cytosol, activation of caspase-3 and OGD time-dependent cell death in WT primary hippocampal neurons. In contrast, in NP1-KO neurons there was no translocation of Bad and Bax from cytosol to the mitochondria, and no evidence of ΔΨ(m) loss, increased Cyt C release and caspase-3 activation following OGD; which resulted in significantly reduced neuronal death. Our results indicate a regulatory role of NP1 in Bad/Bax-dependent mitochondrial release of Cyt C and caspase-3 activation. Together our findings demonstrate a novel mechanism by which NP1 regulates mitochondria-driven hippocampal cell death; suggesting NP1 as a potential therapeutic target against HI brain injury in neonates.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures





References
-
- Armstrong JS, Jones DP. Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. FASEB J. 2002;16:1263–1265. - PubMed
-
- Balduini W, Carloni S, Mazzoni E, Cimino M. New therapeutic strategies in perinatal stroke. Curr Drug Targets CNS Neurol Disord. 2004;3(4):315–23. - PubMed
-
- Banasiak KJ, Xia Y, Haddad GG. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol. 2000;62:215–249. - PubMed
-
- Berbari NF, Bishop GA, Askwith CC, Lewis JS, Mykytyn K. Hippocampal neurons possess primary cilia in culture. J Neurosci Res. 2007;85:1095–1100. - PubMed
-
- Blomgren K, Zhu C, Hallin U, Hagberg H. Mitochondria and ischemic reperfusion damage in the adult and in the developing brain. Biochem Biophys Res Commun. 2003;304(3):551–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
