

Deposition of histone variant H2A.Z within gene bodies regulates responsive genes
- PMID: 23071449
- PMCID: PMC3469445
- DOI: 10.1371/journal.pgen.1002988
Deposition of histone variant H2A.Z within gene bodies regulates responsive genes
Abstract
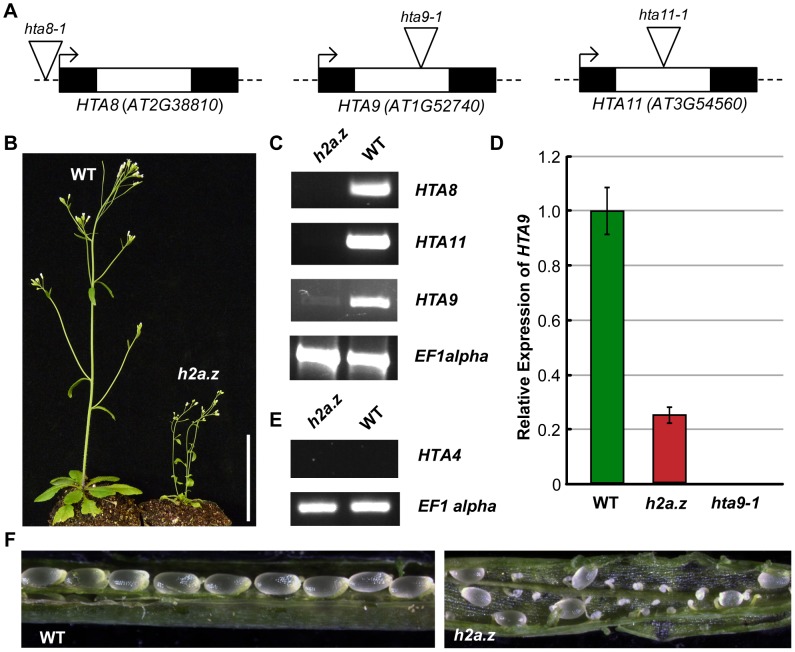
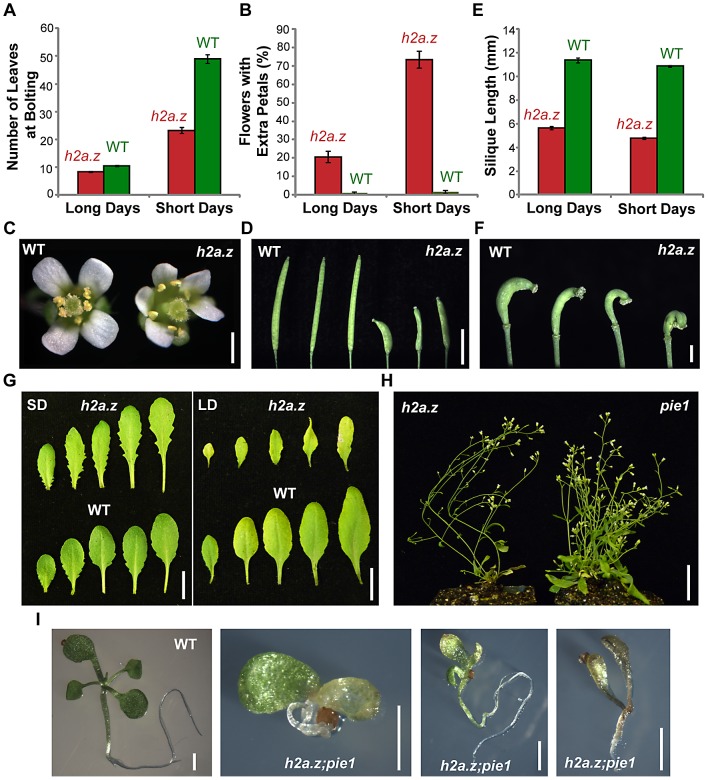
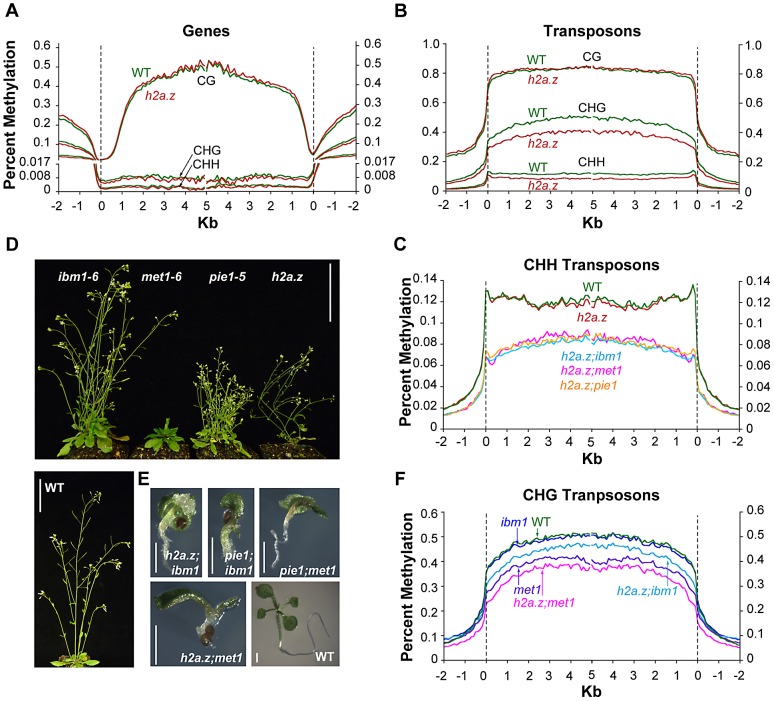
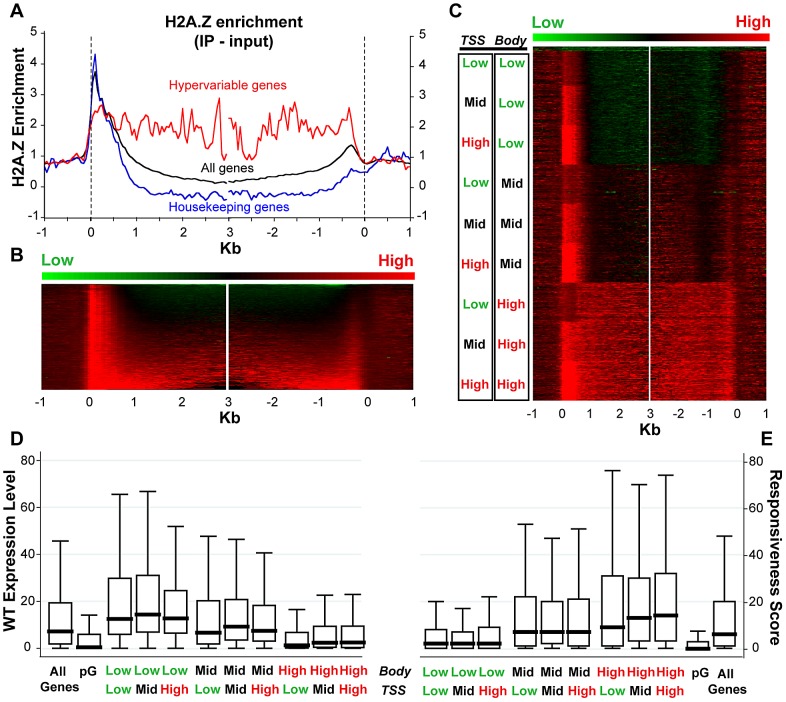
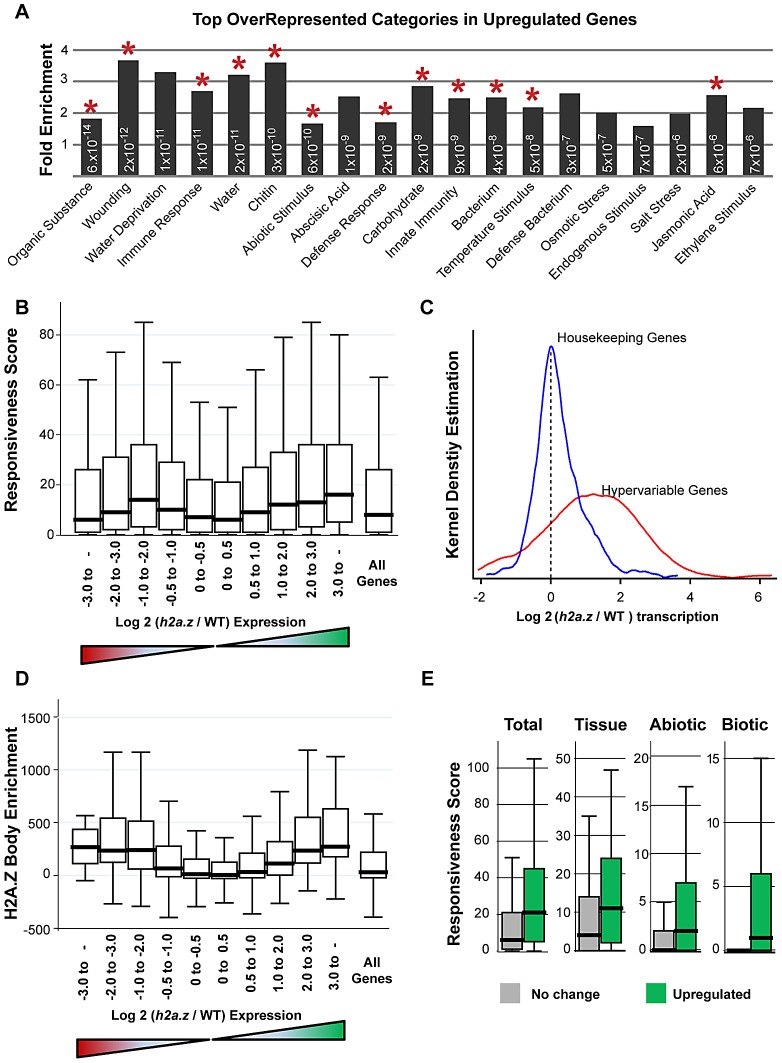
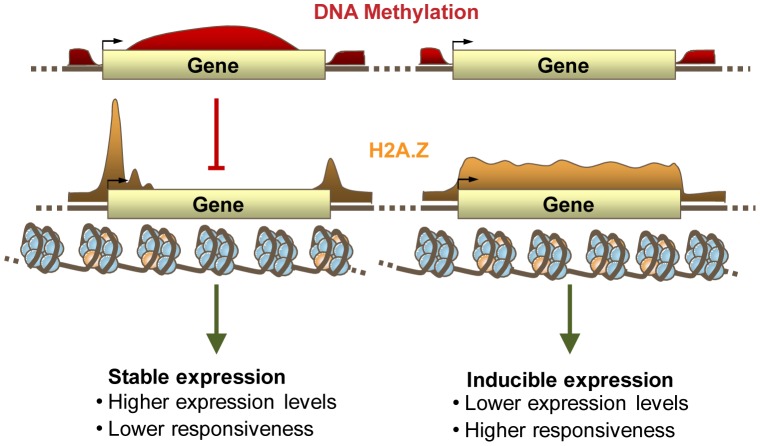
The regulation of eukaryotic chromatin relies on interactions between many epigenetic factors, including histone modifications, DNA methylation, and the incorporation of histone variants. H2A.Z, one of the most conserved but enigmatic histone variants that is enriched at the transcriptional start sites of genes, has been implicated in a variety of chromosomal processes. Recently, we reported a genome-wide anticorrelation between H2A.Z and DNA methylation, an epigenetic hallmark of heterochromatin that has also been found in the bodies of active genes in plants and animals. Here, we investigate the basis of this anticorrelation using a novel h2a.z loss-of-function line in Arabidopsis thaliana. Through genome-wide bisulfite sequencing, we demonstrate that loss of H2A.Z in Arabidopsis has only a minor effect on the level or profile of DNA methylation in genes, and we propose that the global anticorrelation between DNA methylation and H2A.Z is primarily caused by the exclusion of H2A.Z from methylated DNA. RNA sequencing and genomic mapping of H2A.Z show that H2A.Z enrichment across gene bodies, rather than at the TSS, is correlated with lower transcription levels and higher measures of gene responsiveness. Loss of H2A.Z causes misregulation of many genes that are disproportionately associated with response to environmental and developmental stimuli. We propose that H2A.Z deposition in gene bodies promotes variability in levels and patterns of gene expression, and that a major function of genic DNA methylation is to exclude H2A.Z from constitutively expressed genes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Talbert PB, Henikoff S (2010) Histone variants–ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 11: 264–275. - PubMed
-
- Henikoff S, Ahmad K (2005) Assembly of variant histones into chromatin. Annu Rev Cell Dev Biol 21: 133–153. - PubMed
-
- Sarma K, Reinberg D (2005) Histone variants meet their match. Nat Rev Mol Cell Biol 6: 139–149. - PubMed
-
- Zlatanova J, Thakar A (2008) H2A.Z: view from the top. Structure 16: 166–179. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
