

Mismatch repair balances leading and lagging strand DNA replication fidelity
- PMID: 23071460
- PMCID: PMC3469411
- DOI: 10.1371/journal.pgen.1003016
Mismatch repair balances leading and lagging strand DNA replication fidelity
Abstract
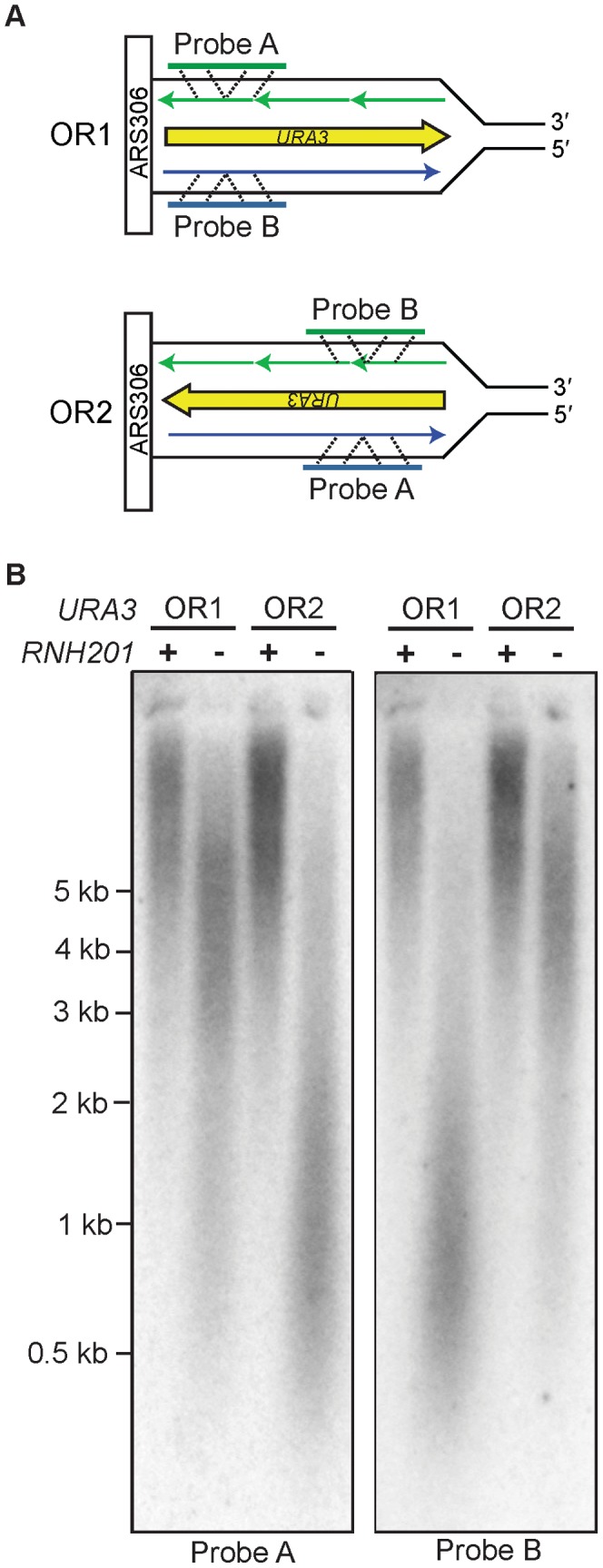
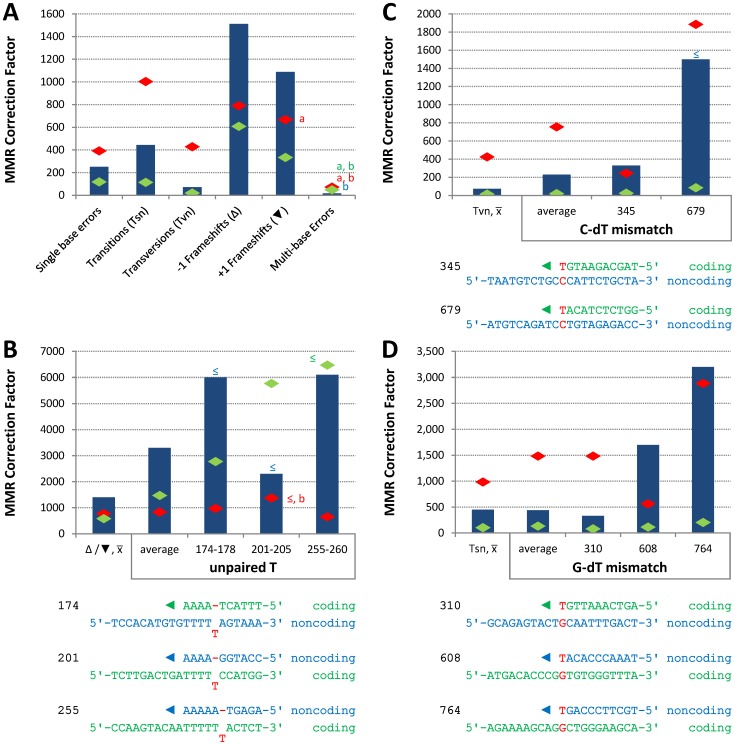
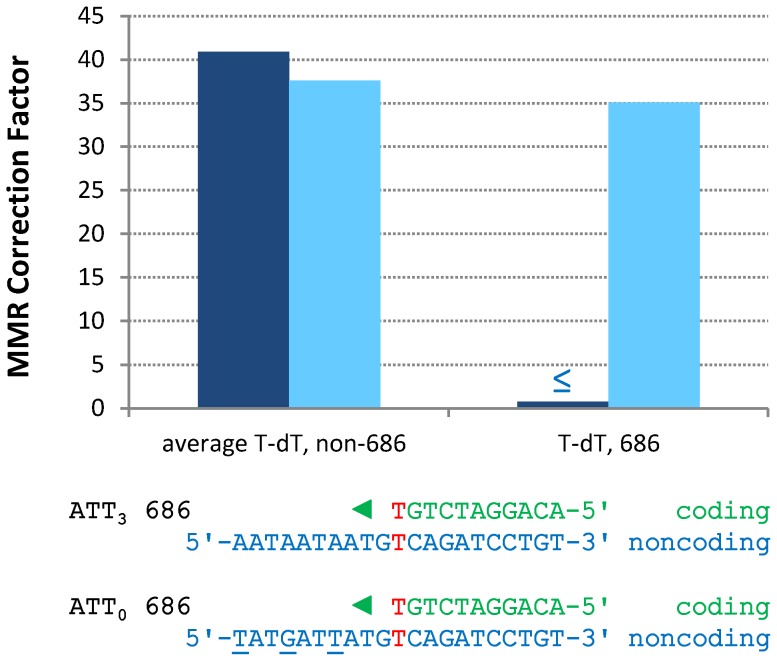
The two DNA strands of the nuclear genome are replicated asymmetrically using three DNA polymerases, α, δ, and ε. Current evidence suggests that DNA polymerase ε (Pol ε) is the primary leading strand replicase, whereas Pols α and δ primarily perform lagging strand replication. The fact that these polymerases differ in fidelity and error specificity is interesting in light of the fact that the stability of the nuclear genome depends in part on the ability of mismatch repair (MMR) to correct different mismatches generated in different contexts during replication. Here we provide the first comparison, to our knowledge, of the efficiency of MMR of leading and lagging strand replication errors. We first use the strand-biased ribonucleotide incorporation propensity of a Pol ε mutator variant to confirm that Pol ε is the primary leading strand replicase in Saccharomyces cerevisiae. We then use polymerase-specific error signatures to show that MMR efficiency in vivo strongly depends on the polymerase, the mismatch composition, and the location of the mismatch. An extreme case of variation by location is a T-T mismatch that is refractory to MMR. This mismatch is flanked by an AT-rich triplet repeat sequence that, when interrupted, restores MMR to > 95% efficiency. Thus this natural DNA sequence suppresses MMR, placing a nearby base pair at high risk of mutation due to leading strand replication infidelity. We find that, overall, MMR most efficiently corrects the most potentially deleterious errors (indels) and then the most common substitution mismatches. In combination with earlier studies, the results suggest that significant differences exist in the generation and repair of Pol α, δ, and ε replication errors, but in a generally complementary manner that results in high-fidelity replication of both DNA strands of the yeast nuclear genome.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
