

Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease
- PMID: 23072311
- PMCID: PMC3971121
- DOI: 10.1146/annurev-pathol-020712-163918
Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease
Abstract
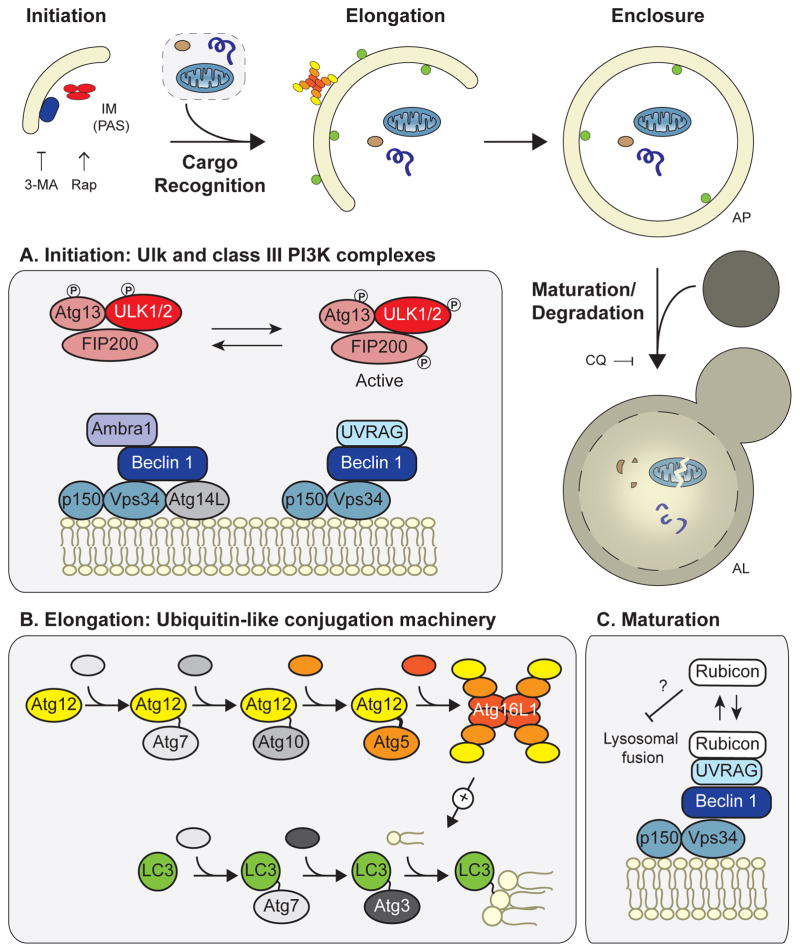
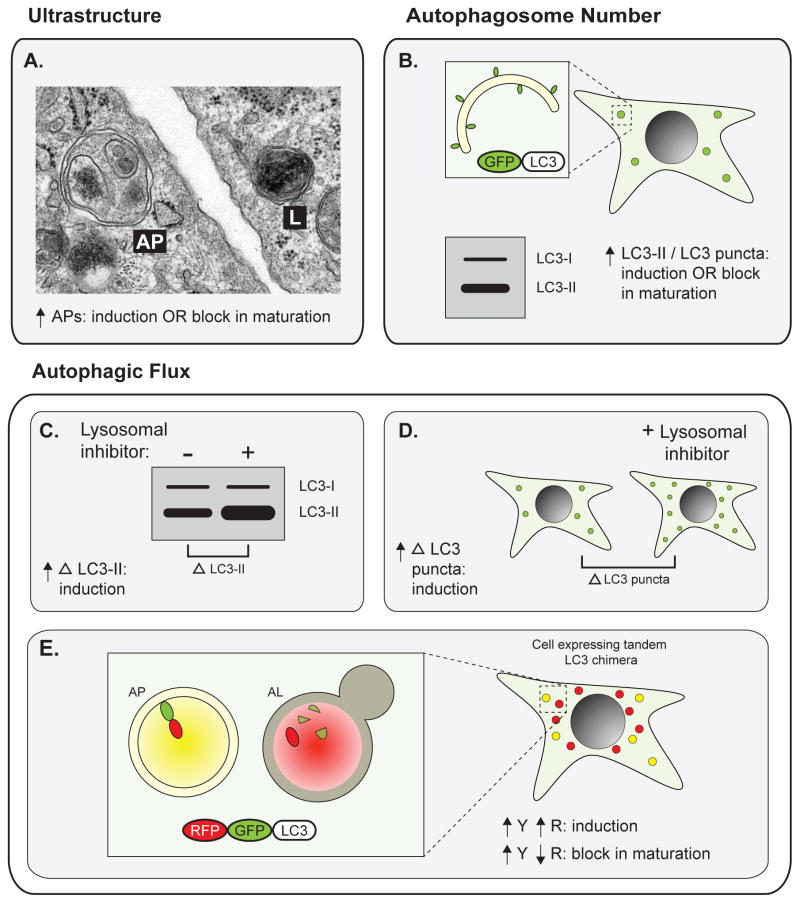
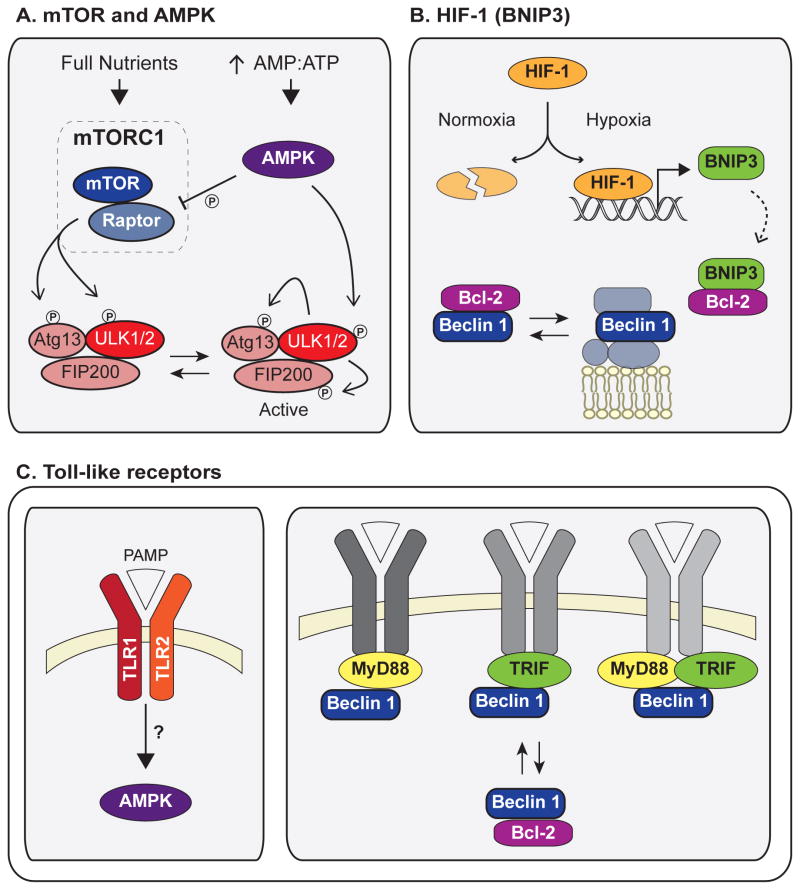
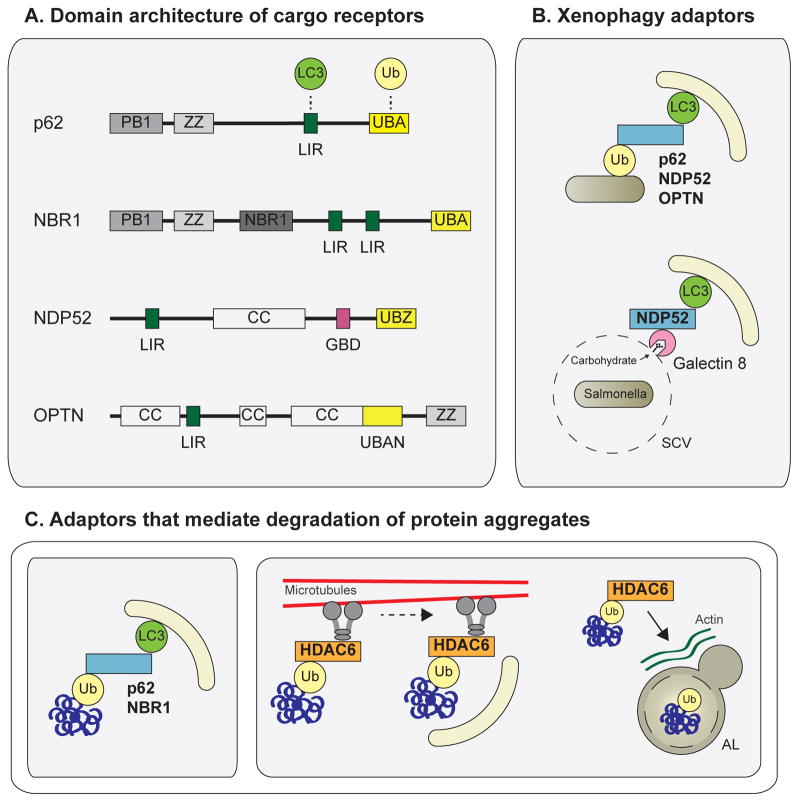
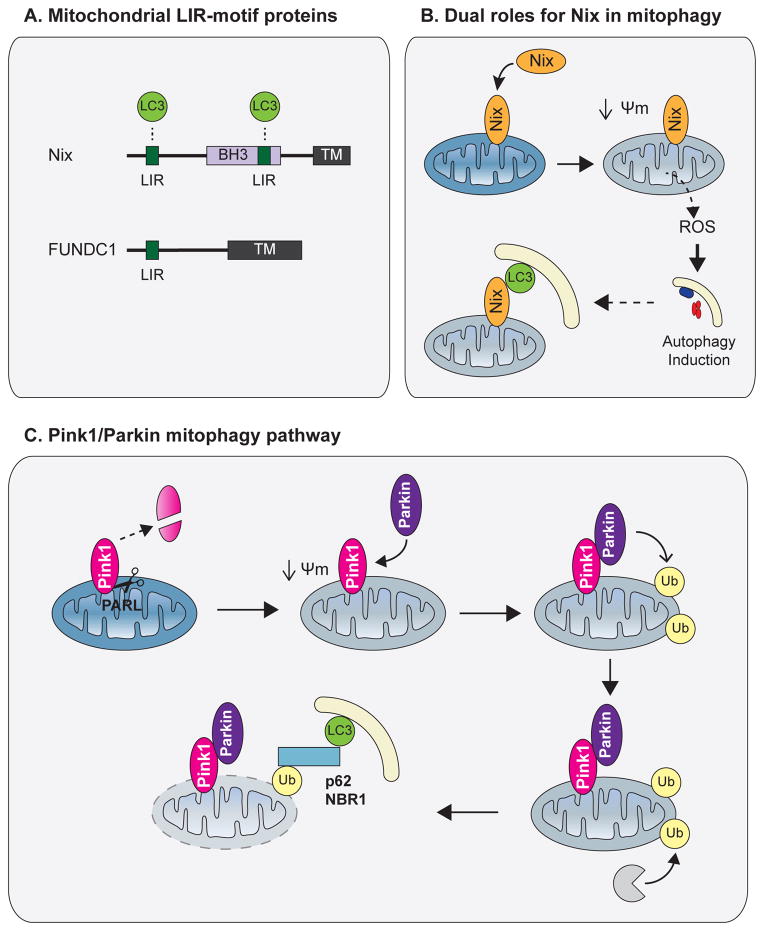
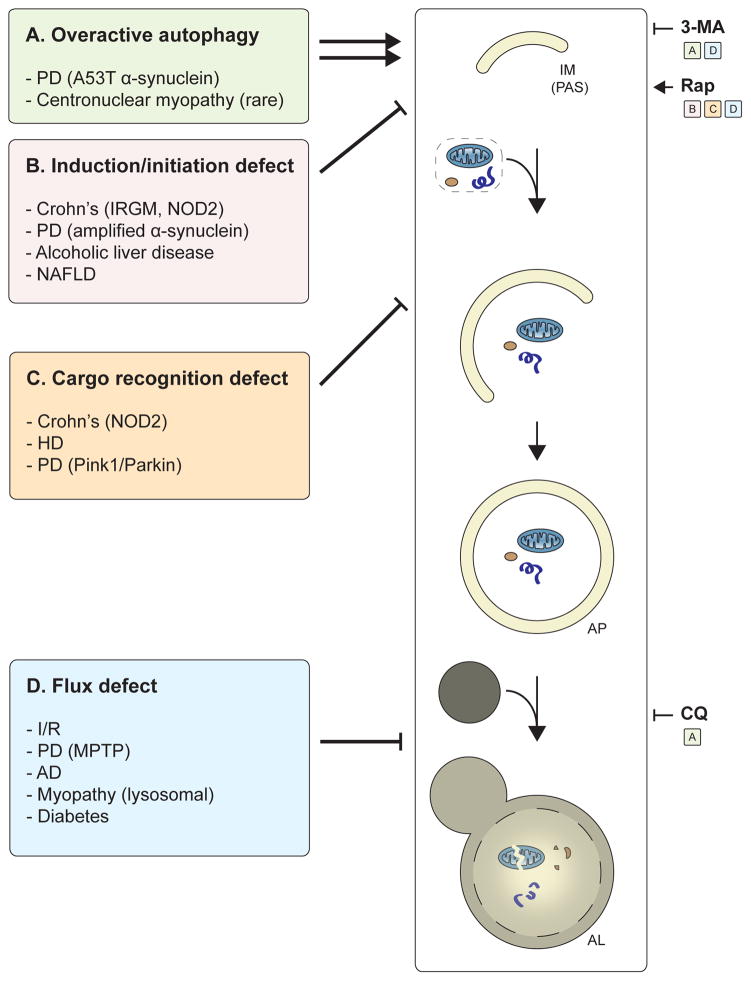
Autophagy, a vital catabolic process that degrades cytoplasmic components within the lysosome, is an essential cytoprotective response to pathologic stresses that occur during diseases such as cancer, ischemia, and infection. In addition to its role as a stress-response pathway, autophagy plays an essential quality-control function in the cell by promoting basal turnover of long-lived proteins and organelles, as well as by selectively degrading damaged cellular components. This homeostatic function protects against a wide variety of diseases, including neurodegeneration, myopathy, liver disease, and diabetes. This review discusses our current understanding of these two principal functions of autophagy and describes in detail how alterations in autophagy promote human disease.
Figures
References
-
- Klionsky DJ, Cregg JM, Dunn WA, Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M. A Unified Nomenclature for Yeast Autophagy-Related Genes. Developmental cell. 2003;5:539–45. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
