

Cardiac structural and sarcomere genes associated with cardiomyopathy exhibit marked intolerance of genetic variation
- PMID: 23074333
- PMCID: PMC3526690
- DOI: 10.1161/CIRCGENETICS.112.963421
Cardiac structural and sarcomere genes associated with cardiomyopathy exhibit marked intolerance of genetic variation
Abstract
Background: The clinical significance of variants in genes associated with inherited cardiomyopathies can be difficult to determine because of uncertainty regarding population genetic variation and a surprising amount of tolerance of the genome even to loss-of-function variants. We hypothesized that genes associated with cardiomyopathy might be particularly resistant to the accumulation of genetic variation.
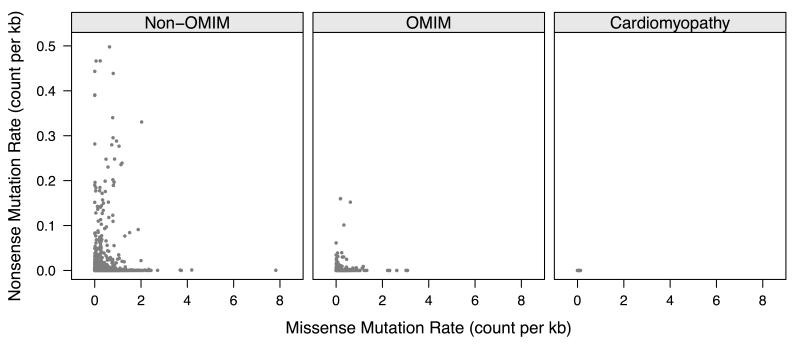
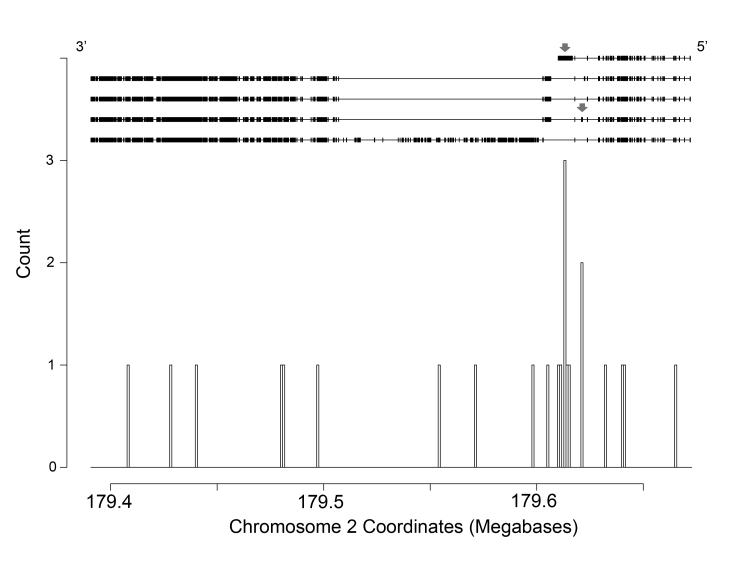
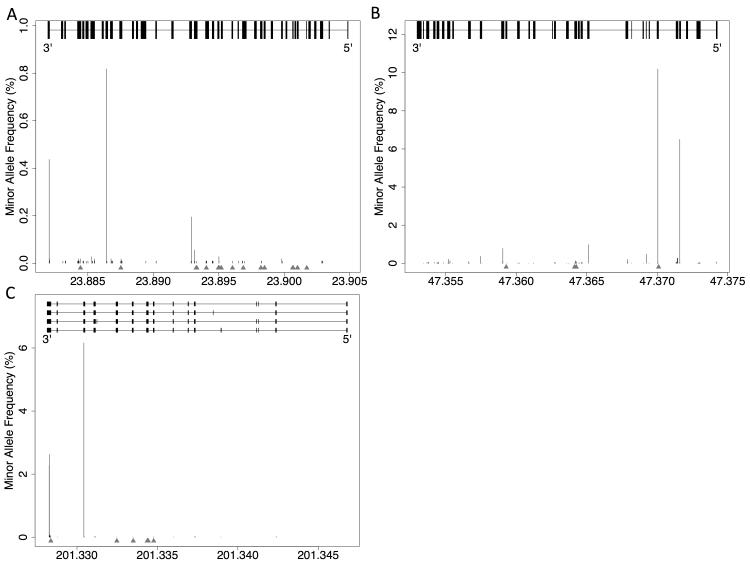
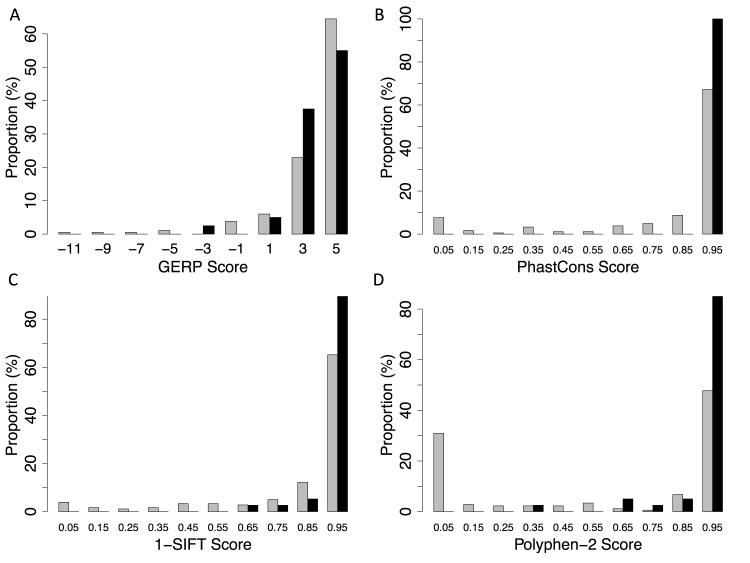
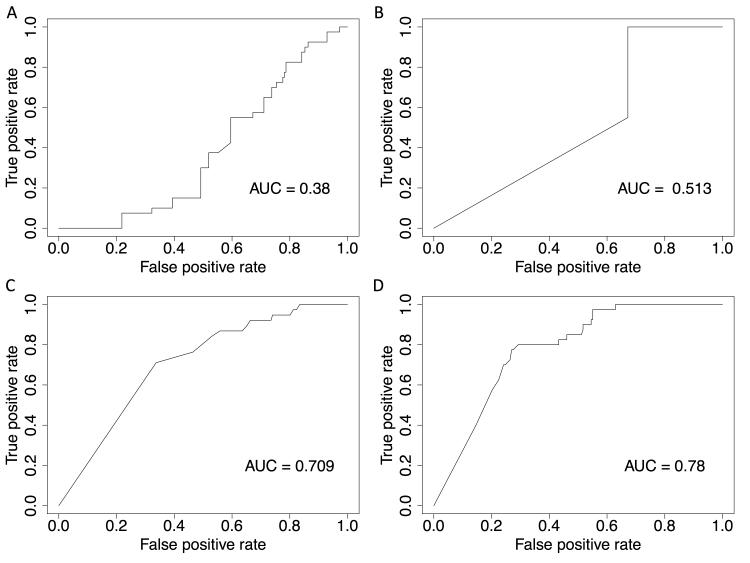
Methods and results: We analyzed the rates of single nucleotide genetic variation in all known genes from the exomes of >5000 individuals from the National Heart, Lung, and Blood Institute's Exome Sequencing Project, as well as the rates of structural variation from the Database of Genomic Variants. Most variants were rare, with over half unique to 1 individual. Cardiomyopathy-associated genes exhibited a rate of nonsense variants, about 96.1% lower than other Mendelian disease genes. We tested the ability of in silico algorithms to distinguish between a set of variants in MYBPC3, MYH7, and TNNT2 with strong evidence for pathogenicity and variants from the Exome Sequencing Project data. Algorithms based on conservation at the nucleotide level (genomic evolutionary rate profiling, PhastCons) did not perform as well as amino acid-level prediction algorithms (Polyphen-2, SIFT). Variants with strong evidence for disease causality were found in the Exome Sequencing Project data at prevalence higher than expected.
Conclusions: Genes associated with cardiomyopathy carry very low rates of population variation. The existence in population data of variants with strong evidence for pathogenicity suggests that even for Mendelian disease genetics, a probabilistic weighting of multiple variants may be preferred over the single gene causality model.
Figures
Comment in
-
Action and the actionability in exome variation.Circ Cardiovasc Genet. 2012 Dec;5(6):597-8. doi: 10.1161/CIRCGENETICS.112.965152. Circ Cardiovasc Genet. 2012. PMID: 23250897 Free PMC article. No abstract available.
References
-
- Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 2010;42:969–972. - PubMed
-
- Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2761–2796. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL-102924/HL/NHLBI NIH HHS/United States
- DP2OD004613/OD/NIH HHS/United States
- HL-102925/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-102923/HL/NHLBI NIH HHS/United States
- DP2 OD006511/OD/NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-103010/HL/NHLBI NIH HHS/United States
- UL1 RR029890/RR/NCRR NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- 5T15LM007033/LM/NLM NIH HHS/United States
- R01HL105993/HL/NHLBI NIH HHS/United States
- R01 HL105993/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- T15 LM007033/LM/NLM NIH HHS/United States
- HL-102926/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UL1RR029890/RR/NCRR NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
