

Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways
- PMID: 23075118
- PMCID: PMC3613173
- DOI: 10.1089/ars.2012.4973
Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways
Abstract
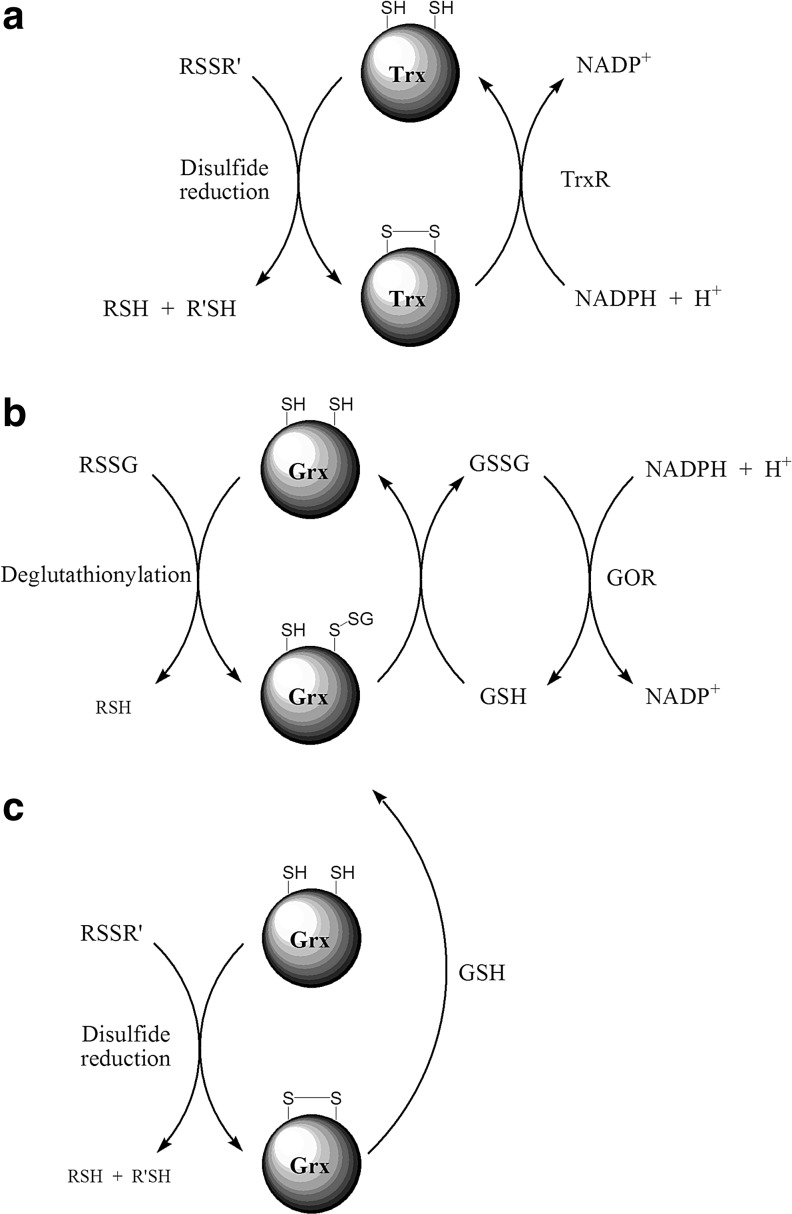
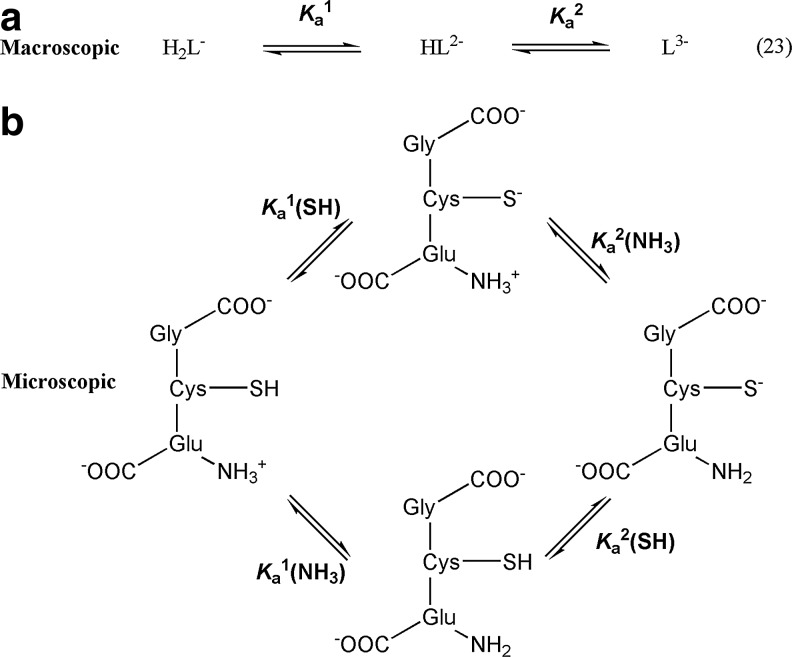
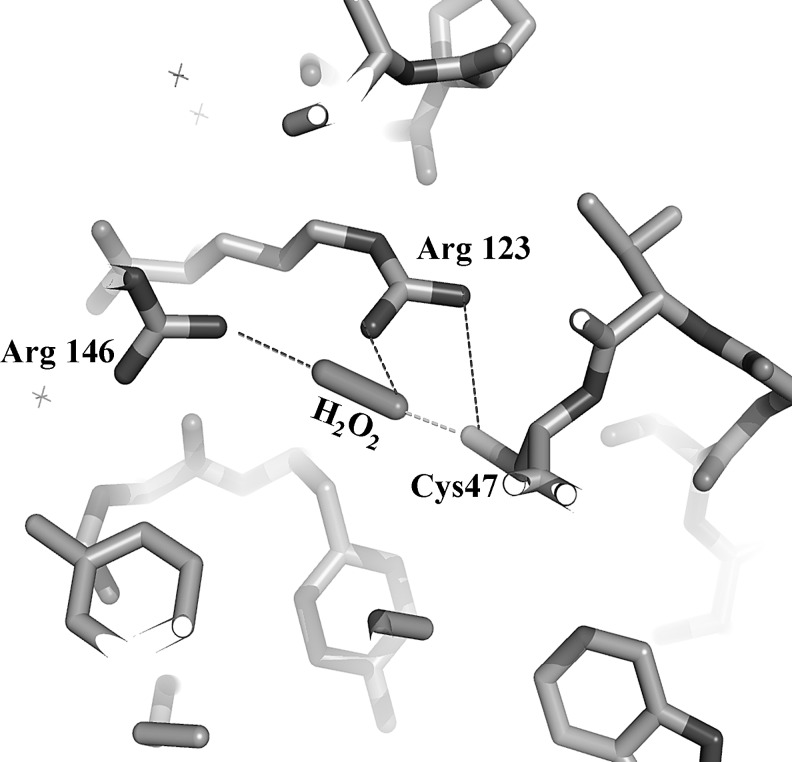
Significance: Disulfides are important building blocks in the secondary and tertiary structures of proteins, serving as inter- and intra-subunit cross links. Disulfides are also the major products of thiol oxidation, a process that has primary roles in defense mechanisms against oxidative stress and in redox regulation of cell signaling. Although disulfides are relatively stable, their reduction, isomerisation, and interconversion as well as their production reactions are catalyzed by delicate enzyme machineries, providing a dynamic system in biology. Redox homeostasis, a thermodynamic parameter that determines which reactions can occur in cellular compartments, is also balanced by the thiol-disulfide pool. However, it is the kinetic properties of the reactions that best represent cell dynamics, because the partitioning of the possible reactions depends on kinetic parameters.
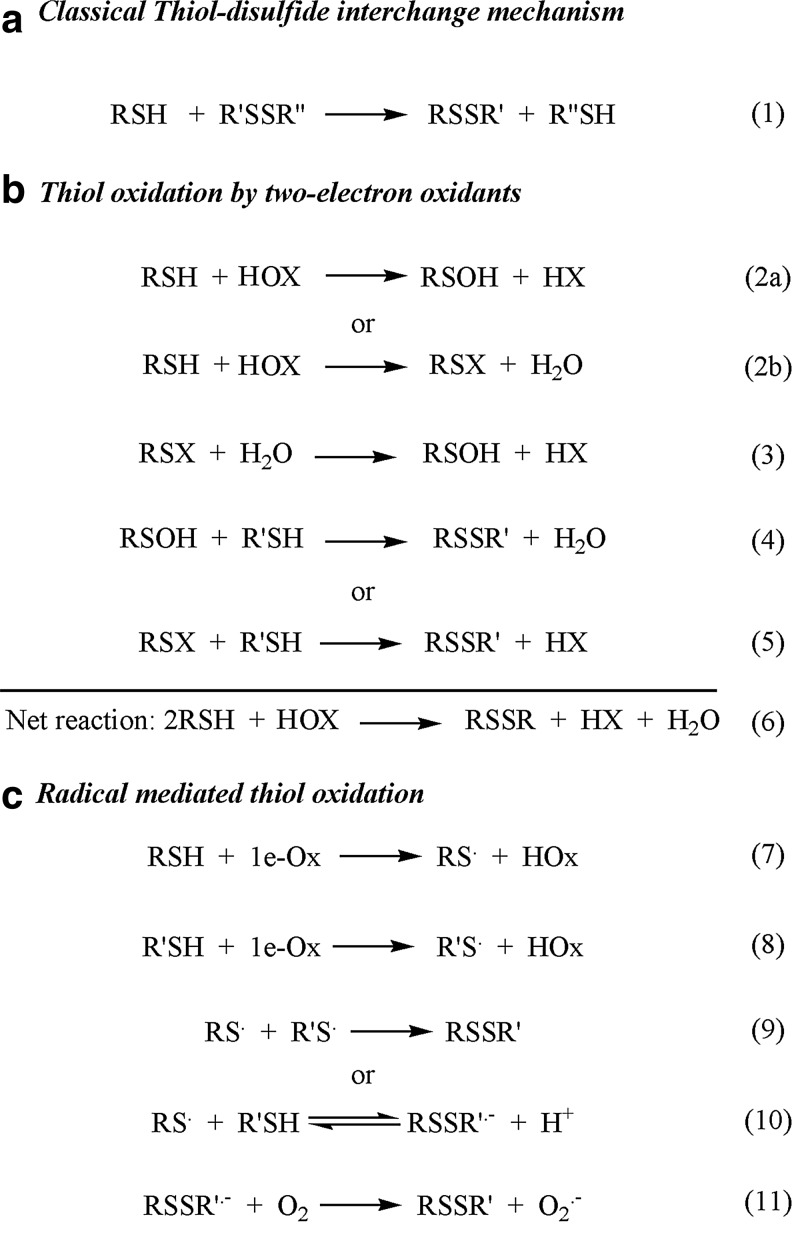
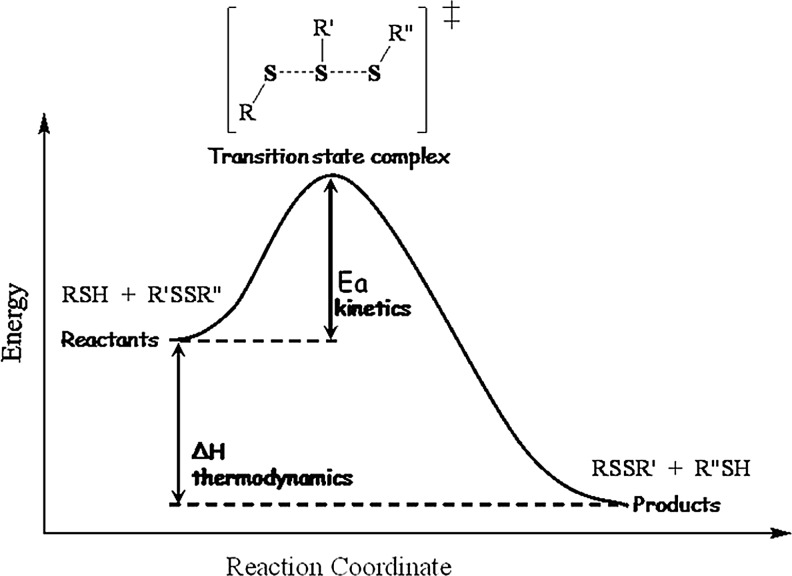
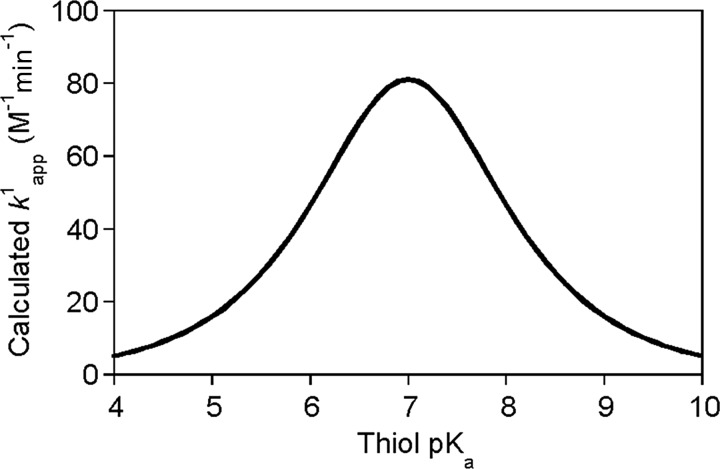
Critical issues: This review is focused on the kinetics and mechanisms of thiol-disulfide substitution and redox reactions. It summarizes the challenges and advances that are associated with kinetic investigations in small molecular and enzymatic systems from a rigorous chemical perspective using biological examples. The most important parameters that influence reaction rates are discussed in detail.
Recent advances and future directions: Kinetic studies of proteins are more challenging than small molecules, and quite often investigators are forced to sacrifice the rigor of the experimental approach to obtain the important kinetic and mechanistic information. However, recent technological advances allow a more comprehensive analysis of enzymatic systems via using the systematic kinetics apparatus that was developed for small molecule reactions, which is expected to provide further insight into the cell's machinery.
Figures
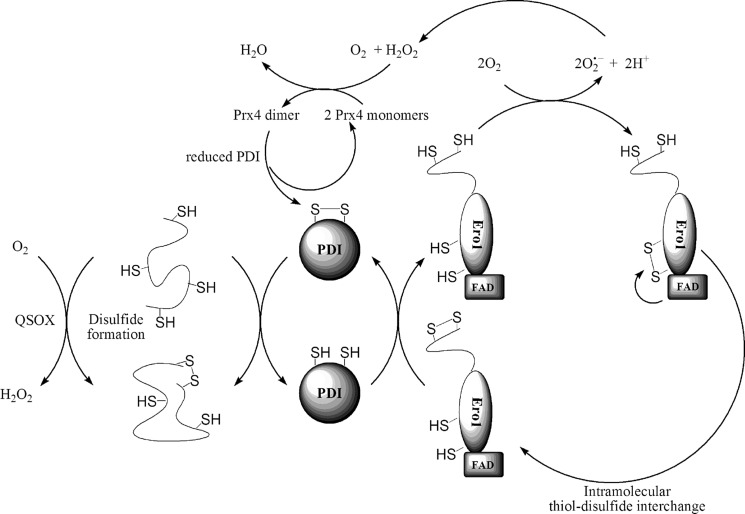
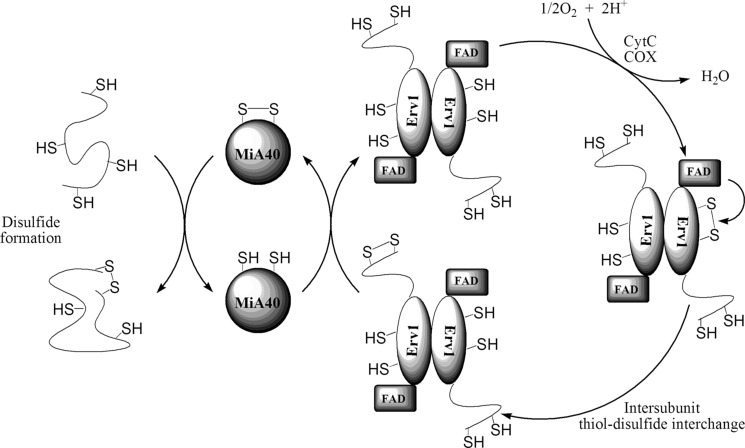
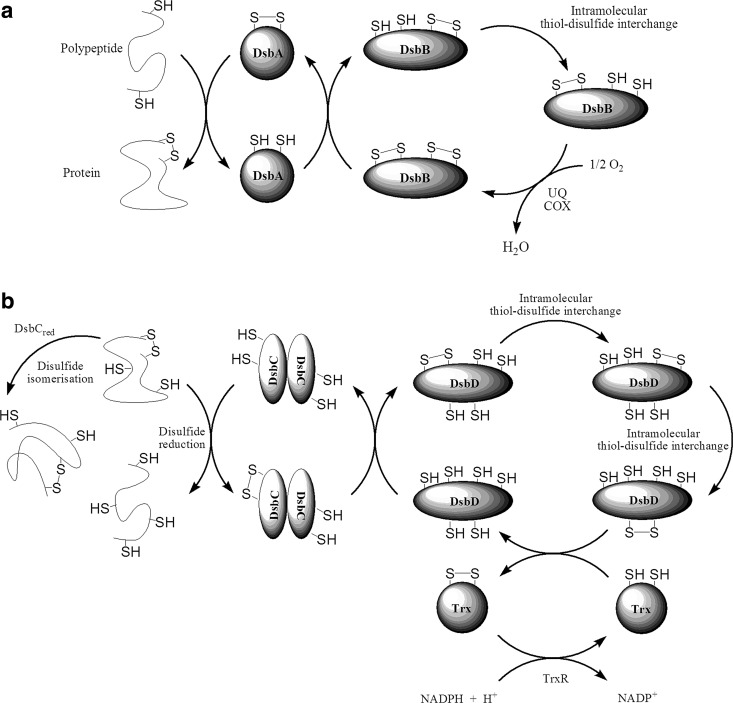
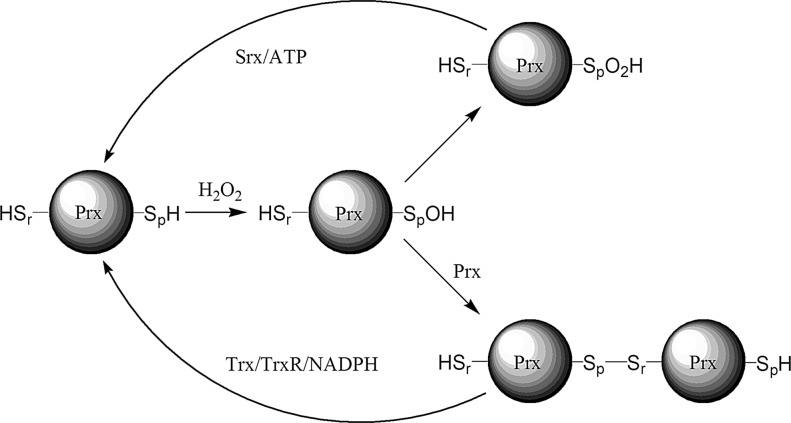
References
-
- Adams GE. McNaught GS. Michael BD. Pulse radiolysis of sulphur compounds 2. Free radical repair by hydrogen transfer from sulphydryl compounds. Trans Farad Soc. 1968;64:902–910.
-
- Ainavarapu SRK. Wiita AP. Dougan L. Uggerud E. Fernandez JM. Single-molecule force spectroscopy measurements of bond elongation during a bimolecular reaction. J Am Chem Soc. 2008;130:6479–6487. - PubMed
-
- Armesto XL. Canle LM. Fernandez MI. Garcia MV. Santaballa JA. First steps in the oxidation of sulfur-containing amino acids by hypohalogenation: very fast generation of intermediate sulfenyl halides and halosulfonium cations. Tetrahedron. 2000;56:1103–1109.
-
- Armstrong DA. Applications of pulse-radiolysis for the study of short-lived sulfur species. In: Chatgilialogy C, editor; Asmas KD, editor. Sulfur-centered reactive intermediates in chemistry and biology. New York: Plenum Press; 1990. pp. 121–134.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
