

Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique
- PMID: 23075513
- PMCID: PMC3528692
- DOI: 10.4161/epi.22552
Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique
Abstract
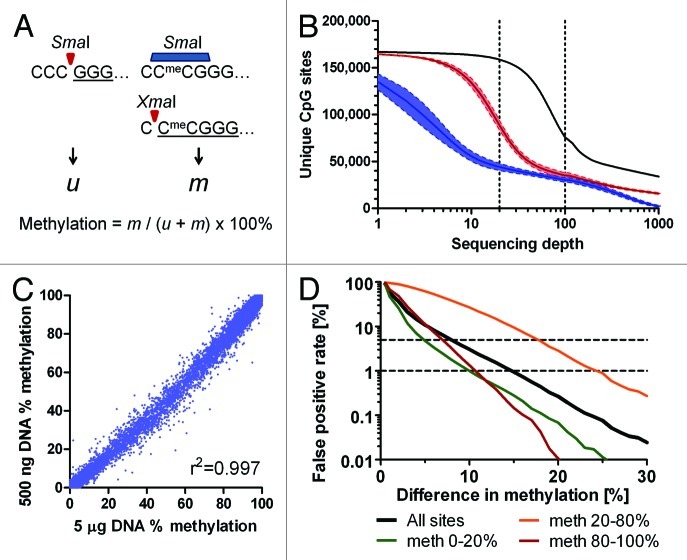
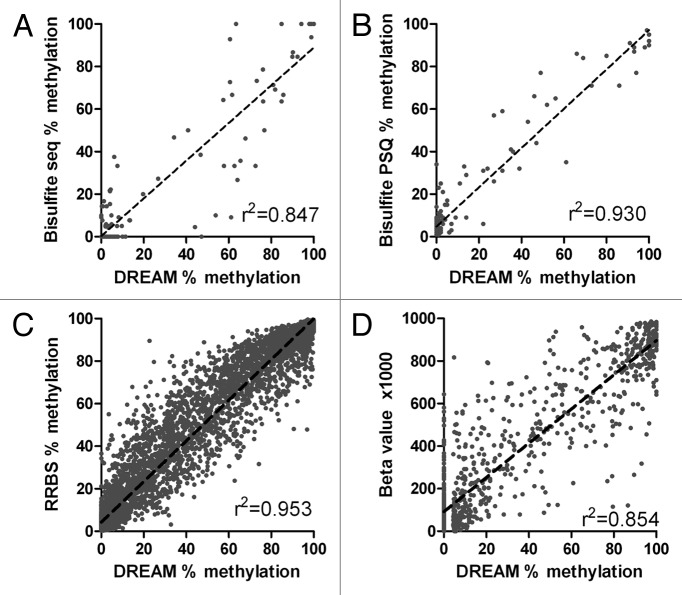
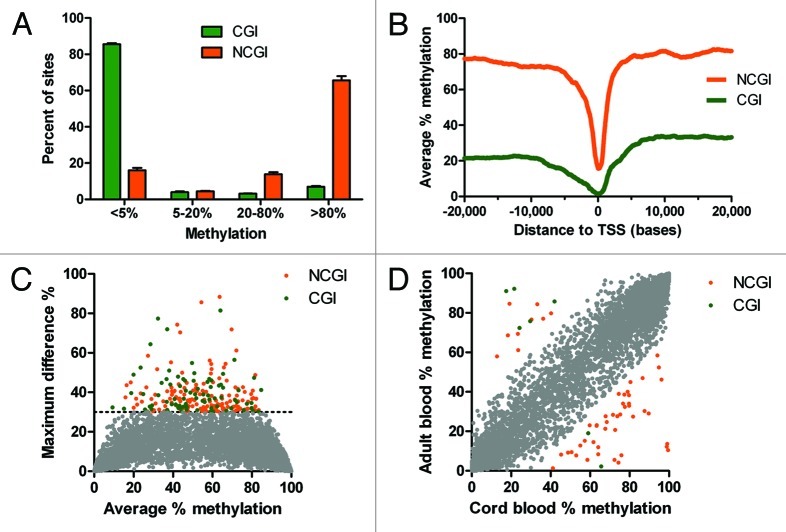
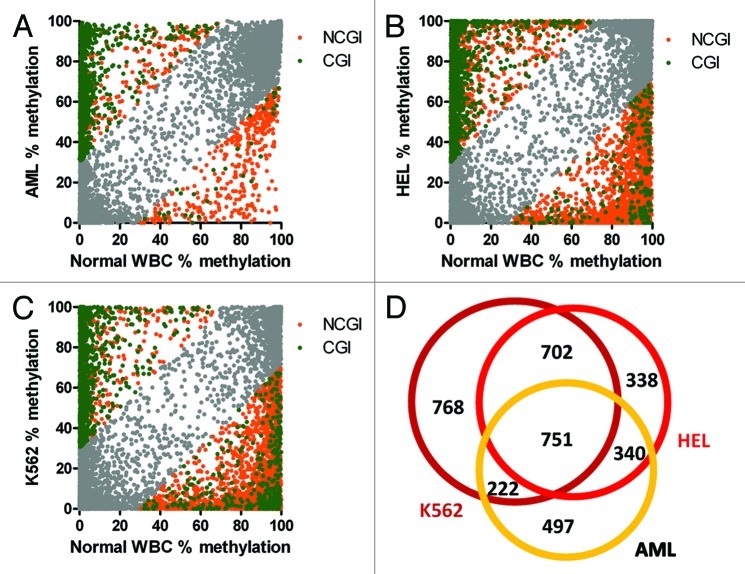
Genome wide analysis of DNA methylation provides important information in a variety of diseases, including cancer. Here, we describe a simple method, Digital Restriction Enzyme Analysis of Methylation (DREAM), based on next generation sequencing analysis of methylation-specific signatures created by sequential digestion of genomic DNA with SmaI and XmaI enzymes. DREAM provides information on 150,000 unique CpG sites, of which 39,000 are in CpG islands and 30,000 are at transcription start sites of 13,000 RefSeq genes. We analyzed DNA methylation in healthy white blood cells and found methylation patterns to be remarkably uniform. Inter individual differences > 30% were observed only at 227 of 28,331 (0.8%) of autosomal CpG sites. Similarly, > 30% differences were observed at only 59 sites when we comparing the cord and adult blood. These conserved methylation patterns contrasted with extensive changes affecting 18-40% of CpG sites in a patient with acute myeloid leukemia and in two leukemia cell lines. The method is cost effective, quantitative (r ( 2) = 0.93 when compared with bisulfite pyrosequencing) and reproducible (r ( 2) = 0.997). Using 100-fold coverage, DREAM can detect differences in methylation greater than 10% or 30% with a false positive rate below 0.05 or 0.001, respectively. DREAM can be useful in quantifying epigenetic effects of environment and nutrition, correlating developmental epigenetic variation with phenotypes, understanding epigenetics of cancer and chronic diseases, measuring the effects of drugs on DNA methylation or deriving new biological insights into mammalian genomes.
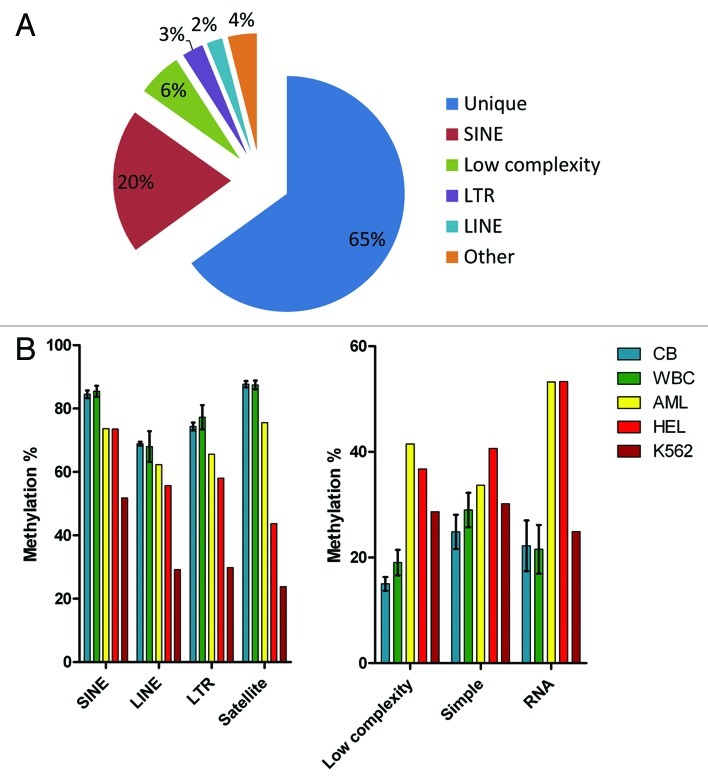
Figures
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- P50 CA100632/CA/NCI NIH HHS/United States
- CA016672/CA/NCI NIH HHS/United States
- CA100632/CA/NCI NIH HHS/United States
- DE022015/DE/NIDCR NIH HHS/United States
- CA123344/CA/NCI NIH HHS/United States
- P01 CA049639/CA/NCI NIH HHS/United States
- K25 CA123344/CA/NCI NIH HHS/United States
- CA121104/CA/NCI NIH HHS/United States
- R01 CA158112/CA/NCI NIH HHS/United States
- CA046939/CA/NCI NIH HHS/United States
- R01 CA121104/CA/NCI NIH HHS/United States
- R01 DE022015/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases