

Interactome-wide prediction of protein-protein binding sites reveals effects of protein sequence variation in Arabidopsis thaliana
- PMID: 23077539
- PMCID: PMC3471968
- DOI: 10.1371/journal.pone.0047022
Interactome-wide prediction of protein-protein binding sites reveals effects of protein sequence variation in Arabidopsis thaliana
Abstract
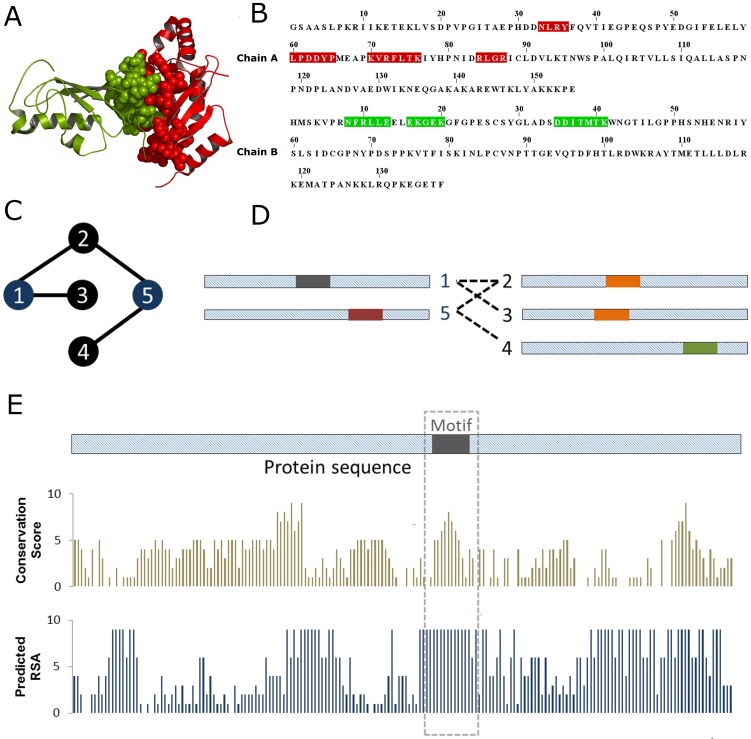
The specificity of protein-protein interactions is encoded in those parts of the sequence that compose the binding interface. Therefore, understanding how changes in protein sequence influence interaction specificity, and possibly the phenotype, requires knowing the location of binding sites in those sequences. However, large-scale detection of protein interfaces remains a challenge. Here, we present a sequence- and interactome-based approach to mine interaction motifs from the recently published Arabidopsis thaliana interactome. The resultant proteome-wide predictions are available via www.ab.wur.nl/sliderbio and set the stage for further investigations of protein-protein binding sites. To assess our method, we first show that, by using a priori information calculated from protein sequences, such as evolutionary conservation and residue surface accessibility, we improve the performance of interface prediction compared to using only interactome data. Next, we present evidence for the functional importance of the predicted sites, which are under stronger selective pressure than the rest of protein sequence. We also observe a tendency for compensatory mutations in the binding sites of interacting proteins. Subsequently, we interrogated the interactome data to formulate testable hypotheses for the molecular mechanisms underlying effects of protein sequence mutations. Examples include proteins relevant for various developmental processes. Finally, we observed, by analysing pairs of paralogs, a correlation between functional divergence and sequence divergence in interaction sites. This analysis suggests that large-scale prediction of binding sites can cast light on evolutionary processes that shape protein-protein interaction networks.
Conflict of interest statement
Figures




References
-
- Janin J, Rodier F (1995) Protein-protein interaction at crystal contacts. Proteins 23: 580–587. - PubMed
-
- Bogan AA, Thorn KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280: 1–9. - PubMed
-
- Moreira IS, Fernandes PA, Ramos MJ (2007) Hot spots–a review of the protein-protein interface determinant amino-acid residues. Proteins 68: 803–812. - PubMed
-
- de Vries SJ, Bonvin AM (2008) How proteins get in touch: interface prediction in the study of biomolecular complexes. Curr Protein Pept Sci 9: 394–406. - PubMed
-
- Morsy M, Gouthu S, Orchard S, Thorneycroft D, Harper JF, et al. (2008) Charting plant interactomes: possibilities and challenges. Trends Plant Sci 13: 183–191. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
