

On the nature of mycobacteriophage diversity and host preference
- PMID: 23084079
- PMCID: PMC3518647
- DOI: 10.1016/j.virol.2012.09.026
On the nature of mycobacteriophage diversity and host preference
Abstract
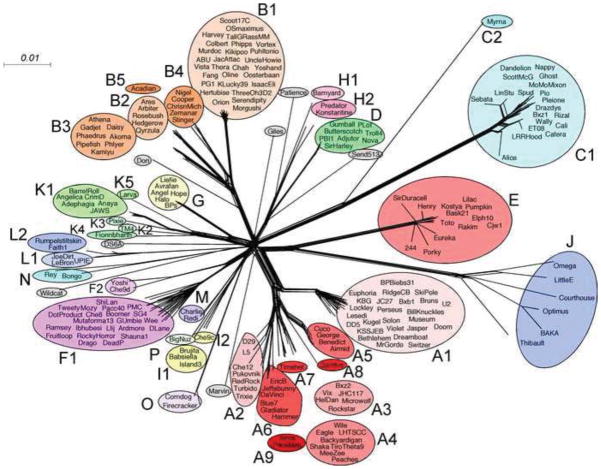
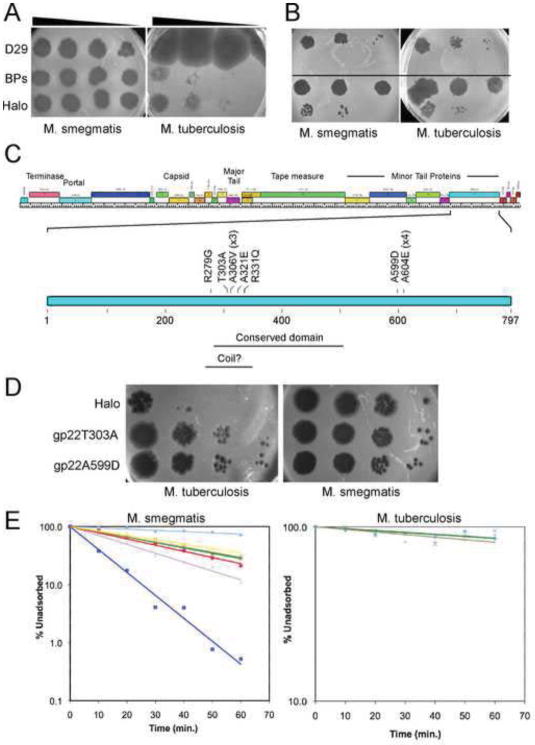
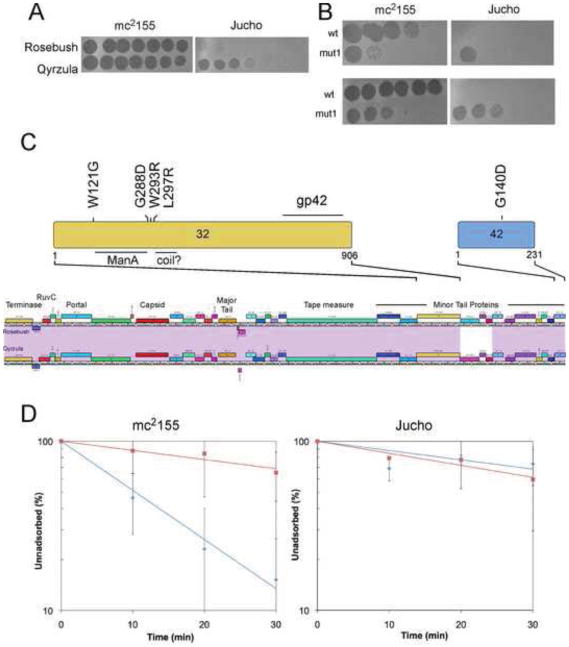
The complete genome sequences of over 220 mycobacteriophages reveal them to be highly diverse, with numerous types sharing little or no nucleotide sequence identity with each other. We have determined the preferences of these phages for Mycobacterium tuberculosis and for other strains of Mycobacterium smegmatis, and find there is a correlation between genome type (cluster, subcluster, singleton) and host range. For many of the phages, expansion of host range occurs at relatively high frequencies, and we describe several examples in which host constraints occur at early stages of infection (adsorption or DNA injection), and phages have the ability to expand their host range through mutations in tail genes. We present a model in which phage diversity is a function of both the ability of phages to rapidly adapt to new hosts and the richness of the diversity of the bacterial population from which those phages are isolated.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures



References
-
- Abrescia NG, Bamford DH, Grimes JM, Stuart DI. Structure unifies the viral universe. Annu Rev Biochem. 2012;81:795–822. - PubMed
-
- Arisaka F, Kanamaru S, Leiman P, Rossmann MG. The tail lysozyme complex of bacteriophage T4. Int J Biochem Cell Biol. 2003;35:16–21. - PubMed
-
- Bae B, Ohene-Adjei S, Kocherginskaya S, Mackie RI, Spies MA, Cann IK, Nair SK. Molecular basis for the selectivity and specificity of ligand recognition by the family 16 carbohydrate-binding modules from Thermoanaerobacterium polysaccharolyticum ManA. J Biol Chem. 2008;283:12415–12425. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
