

CIKS (Act1 or TRAF3IP2) mediates high glucose-induced endothelial dysfunction
- PMID: 23085260
- PMCID: PMC3606809
- DOI: 10.1016/j.cellsig.2012.10.009
CIKS (Act1 or TRAF3IP2) mediates high glucose-induced endothelial dysfunction
Abstract
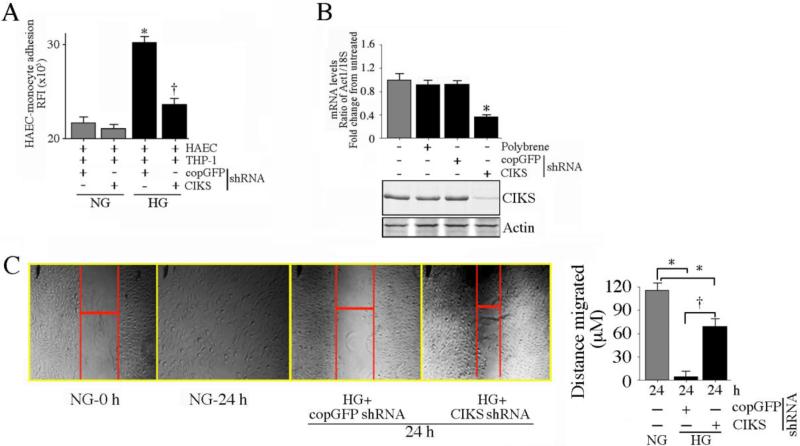
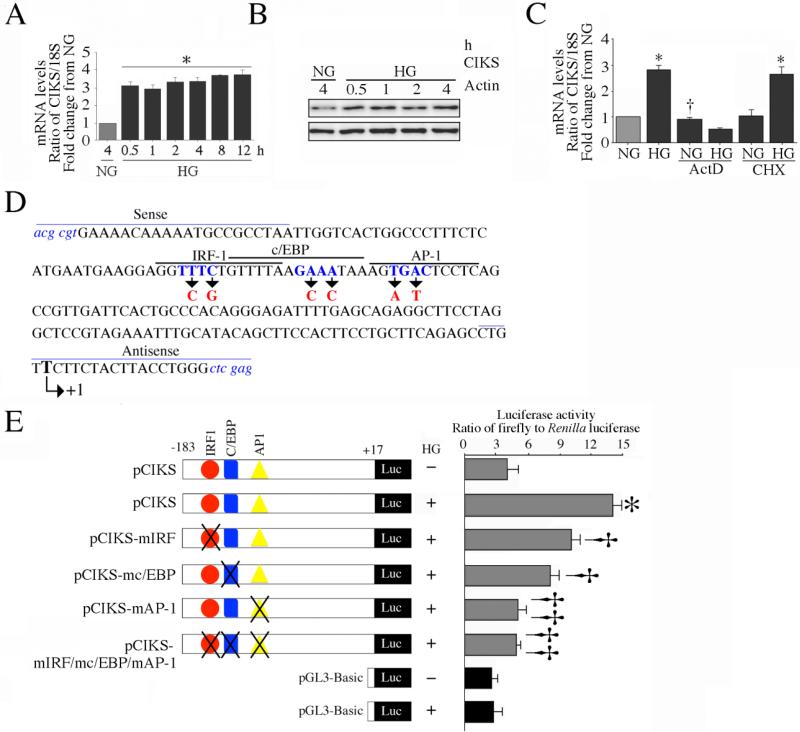
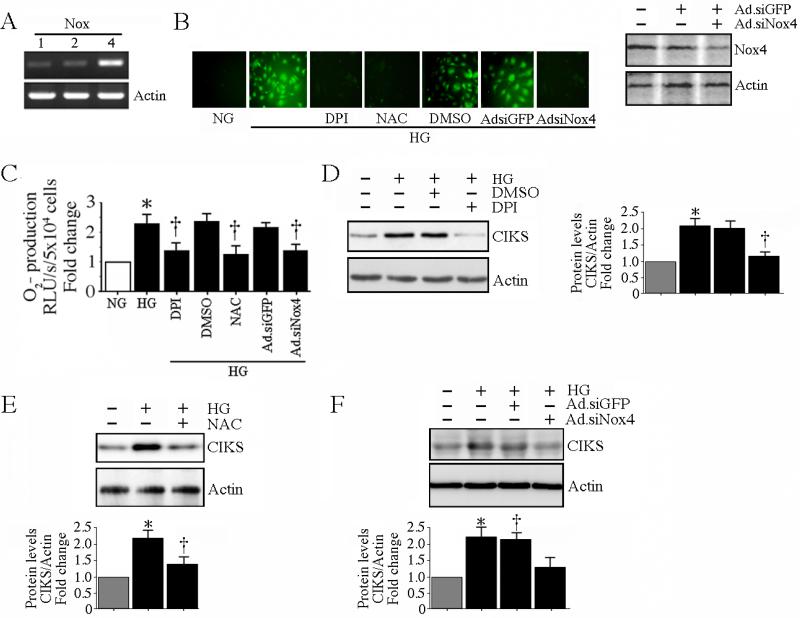
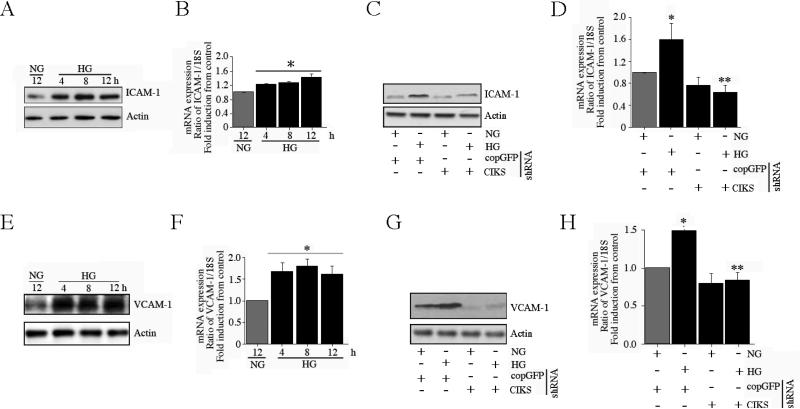
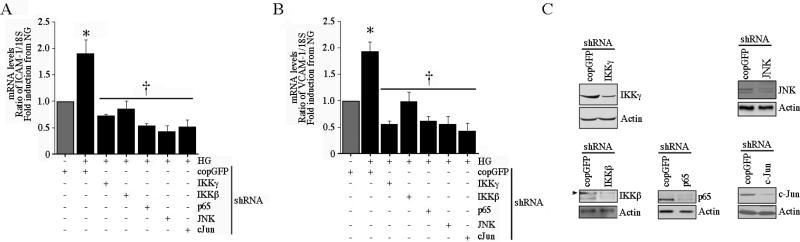
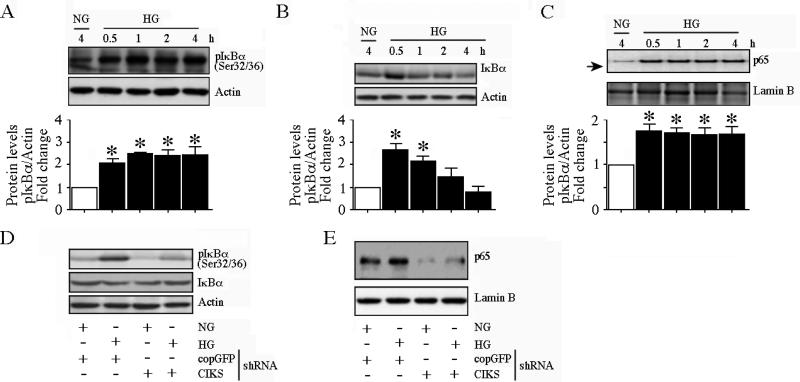
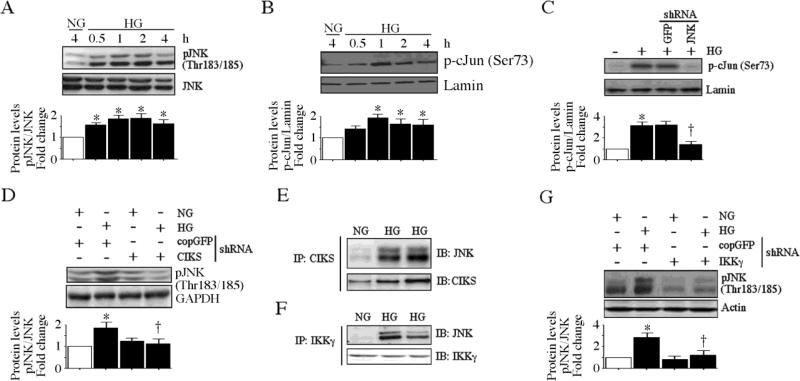
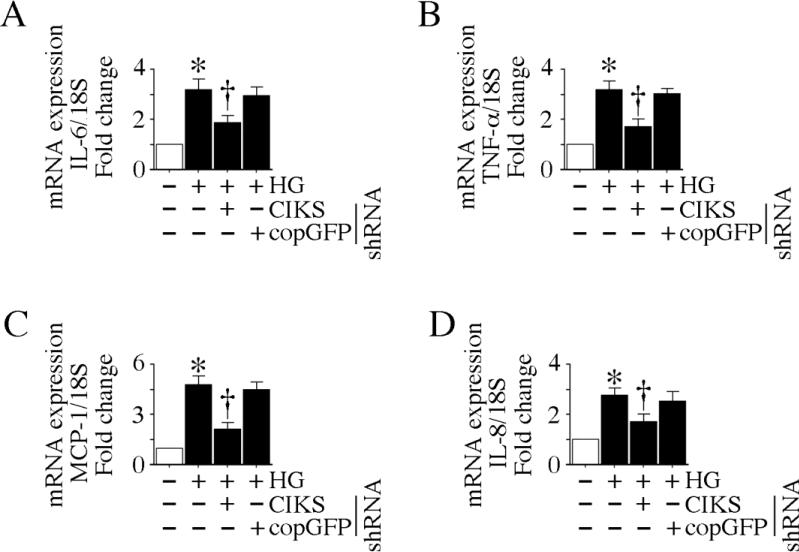
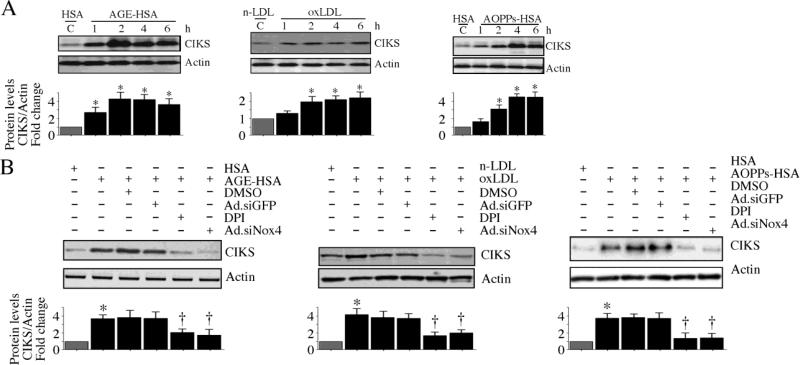
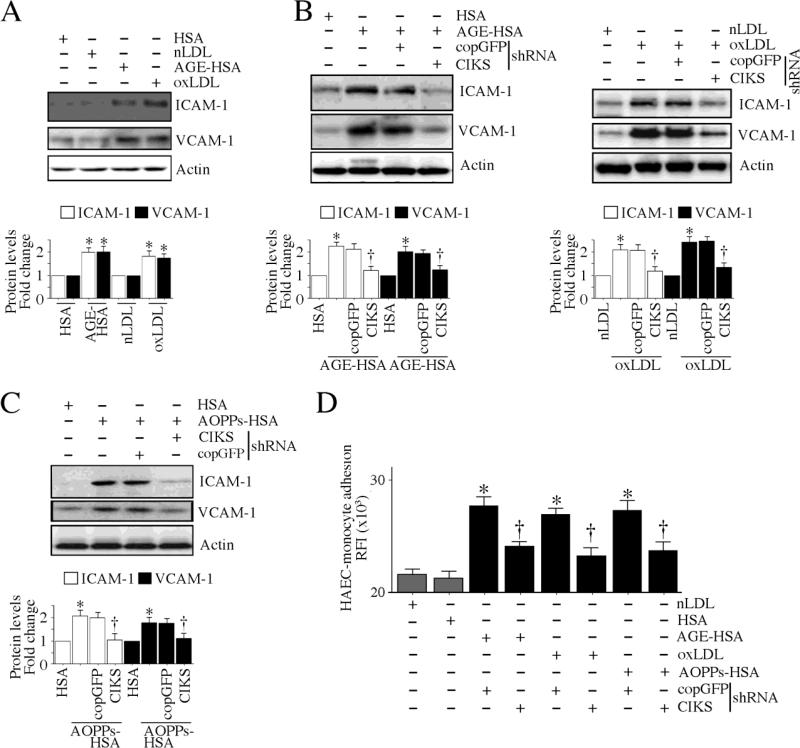
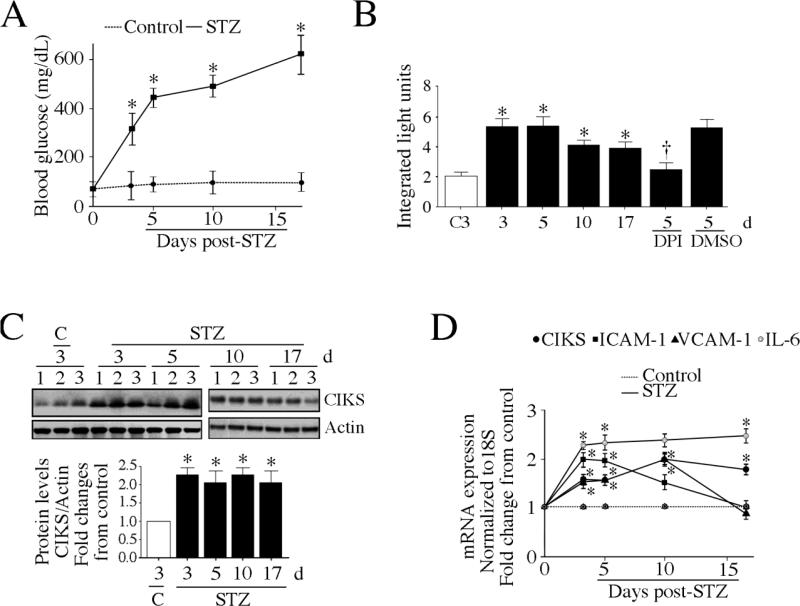
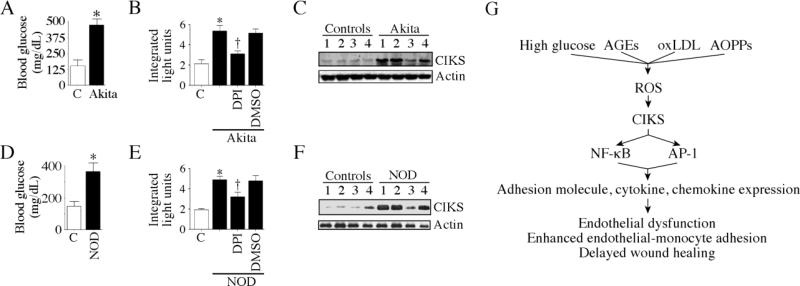
Hyperglycemia-induced endothelial dysfunction is characterized by enhanced inflammatory cytokine and adhesion molecule expression, and endothelial-monocyte adhesion. The adapter molecule CIKS (connection to IKK and SAPK/JNK; also known as Act1 or TRAF3IP2) is an upstream regulator of NF-κB and AP-1, and plays a role in inflammation and injury. Here we show that high glucose (HG; 25mM vs. 5mM d-glucose)-induced endothelial-monocyte adhesion and inhibition of endothelial cell (EC) migration were both reversed by CIKS knockdown. In EC, HG induced CIKS mRNA and protein expression via DPI-inhibitable Nox4-dependent ROS generation. Further, HG induced CIKS transcription and enhanced CIKS promoter-dependent reporter gene activation via Nox4, ROS, AP-1 and C/EBP. Coimmunoprecipitation and immunoblotting revealed CIKS/IKKβ/JNK physical association under basal conditions that was enhanced by HG treatment. Importantly, CIKS knockdown inhibited HG-induced (i) IKKβ and JNK phosphorylation, (ii) p65 and c-Jun nuclear translocation, and (iii) NF-κB- and AP-1-dependent proinflammatory cytokine, chemokine, and adhesion molecule expression. Similar to HG, the deleterious metabolic products of chronic hyperglycemia, AGE-HSA, AOPPs-HSA and oxLDL, also induced CIKS-dependent endothelial dysfunction. Notably, aortas from streptozotocin-induced and the autoimmune type 1 diabetic NOD and Akita mice showed enhanced DPI-inhibitable ROS generation and CIKS expression. Since CIKS mediates high glucose-induced NF-κB and AP-1-dependent inflammatory signaling and endothelial dysfunction, targeting CIKS may delay progression of vascular diseases during diabetes mellitus and atherosclerosis.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
CIKS (Act1 or TRAF3IP2) mediates Angiotensin-II-induced Interleukin-18 expression, and Nox2-dependent cardiomyocyte hypertrophy.J Mol Cell Cardiol. 2012 Jul;53(1):113-24. doi: 10.1016/j.yjmcc.2012.04.009. Epub 2012 Apr 26. J Mol Cell Cardiol. 2012. PMID: 22575763 Free PMC article.
-
TRAF3IP2 mediates high glucose-induced endothelin-1 production as well as endothelin-1-induced inflammation in endothelial cells.Am J Physiol Heart Circ Physiol. 2018 Jan 1;314(1):H52-H64. doi: 10.1152/ajpheart.00478.2017. Epub 2017 Sep 29. Am J Physiol Heart Circ Physiol. 2018. PMID: 28971844 Free PMC article.
-
TRAF3IP2 mediates interleukin-18-induced cardiac fibroblast migration and differentiation.Cell Signal. 2013 Nov;25(11):2176-84. doi: 10.1016/j.cellsig.2013.07.013. Epub 2013 Jul 18. Cell Signal. 2013. PMID: 23872479 Free PMC article.
-
High glucose conditions induce upregulation of fractalkine and monocyte chemotactic protein-1 in human smooth muscle cells.Thromb Haemost. 2008 Dec;100(6):1155-65. Thromb Haemost. 2008. PMID: 19132243
-
The anti-inflammatory properties of vinpocetine mediates its therapeutic potential in management of atherosclerosis.J Inflamm (Lond). 2024 Jun 10;21(1):19. doi: 10.1186/s12950-024-00394-x. J Inflamm (Lond). 2024. PMID: 38858751 Free PMC article. Review.
Cited by
-
Identification of potential markers for type 2 diabetes mellitus via bioinformatics analysis.Mol Med Rep. 2020 Sep;22(3):1868-1882. doi: 10.3892/mmr.2020.11281. Epub 2020 Jun 26. Mol Med Rep. 2020. PMID: 32705173 Free PMC article.
-
Anti-inflammatory therapy of atherosclerosis: focusing on IKKβ.J Inflamm (Lond). 2023 Feb 23;20(1):8. doi: 10.1186/s12950-023-00330-5. J Inflamm (Lond). 2023. PMID: 36823573 Free PMC article. Review.
-
Dynamic cellular and molecular modulations of diabetes mediated head and neck carcinogenesis.Oncotarget. 2015 Oct 6;6(30):29268-84. doi: 10.18632/oncotarget.4922. Oncotarget. 2015. PMID: 26337468 Free PMC article.
-
Anti-inflammatory Effects of Atorvastatin in Human Glioblastoma Spheroids Cultured in a Three-dimensional Model: Possible Relevance to Glioblastoma Treatment.Mol Neurobiol. 2018 Mar;55(3):2102-2110. doi: 10.1007/s12035-017-0445-2. Epub 2017 Mar 10. Mol Neurobiol. 2018. PMID: 28283885
-
TRAF3IP2 mediates atherosclerotic plaque development and vulnerability in ApoE(-/-) mice.Atherosclerosis. 2016 Sep;252:153-160. doi: 10.1016/j.atherosclerosis.2016.05.029. Epub 2016 May 19. Atherosclerosis. 2016. PMID: 27237075 Free PMC article.
References
-
- Diabetes Care. 2008;31(Suppl 1):S55–60. - PubMed
-
- Collier A, Small M. Br J Hosp Med. 1991;45(1):38–40. - PubMed
-
- Kalousova M, Skrha J, Zima T. Physiol Res. 2002;51(6):597–604. - PubMed
-
- Martin-Gallan P, Carrascosa A, Gussinye M, Dominguez C. Free Radic Biol Med. 2003;34(12):1563–1574. - PubMed
-
- Boullier A, Bird DA, Chang MK, Dennis EA, Friedman P, Gillotre-Taylor K, Horkko S, Palinski W, Quehenberger O, Shaw P, Steinberg D, Terpstra V, Witztum JL. Ann N Y Acad Sci. 2001;947:214–222. discussion 222-213. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
