

The hereditary spastic paraplegia protein strumpellin: characterisation in neurons and of the effect of disease mutations on WASH complex assembly and function
- PMID: 23085491
- PMCID: PMC3714738
- DOI: 10.1016/j.bbadis.2012.10.011
The hereditary spastic paraplegia protein strumpellin: characterisation in neurons and of the effect of disease mutations on WASH complex assembly and function
Abstract
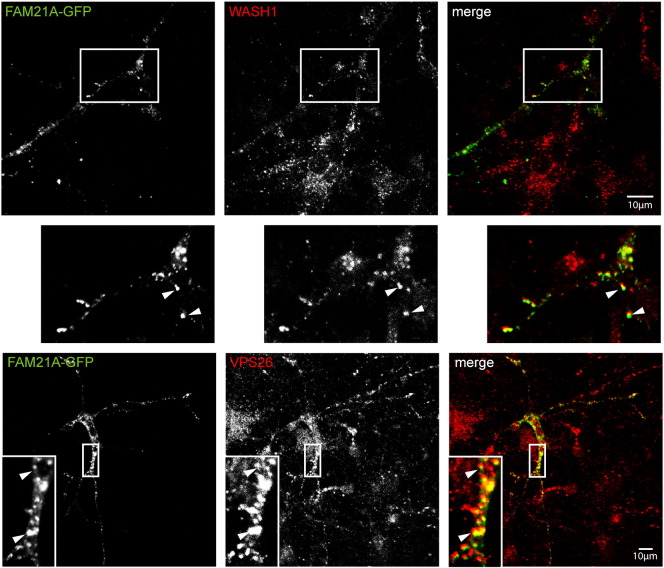
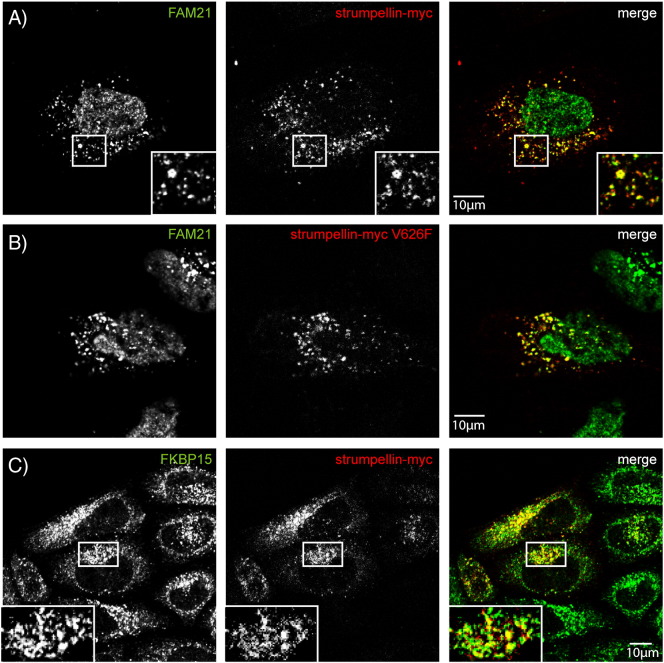
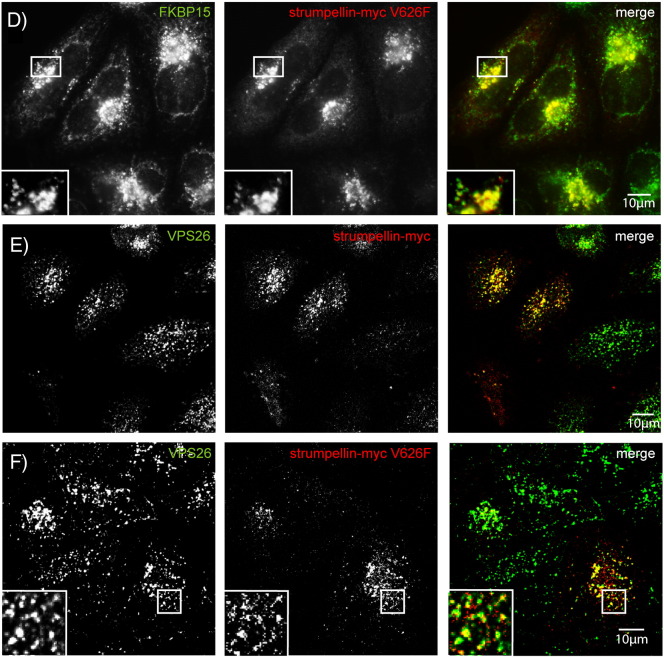
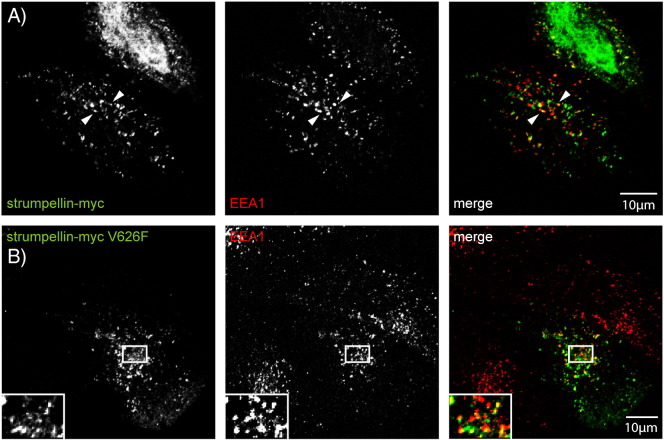
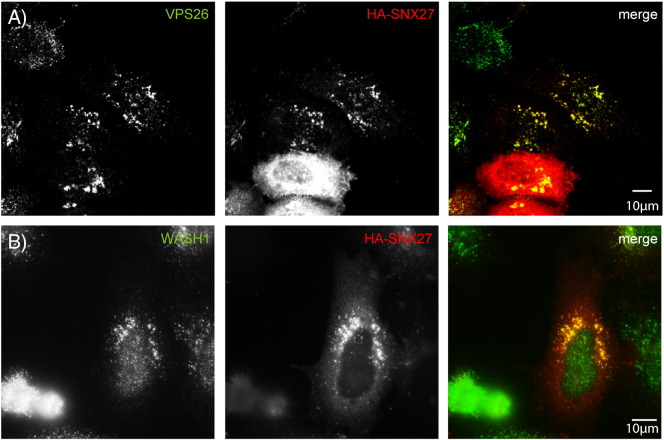
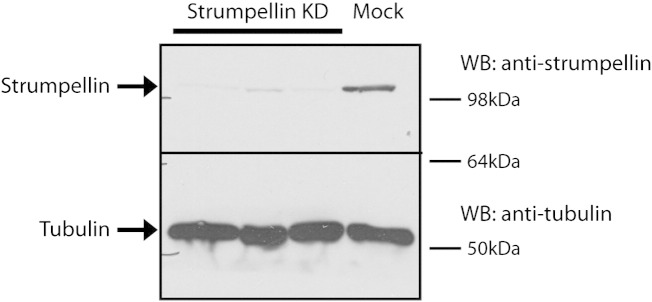
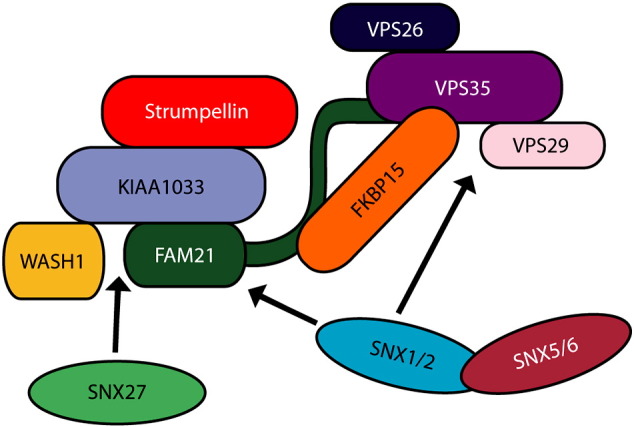
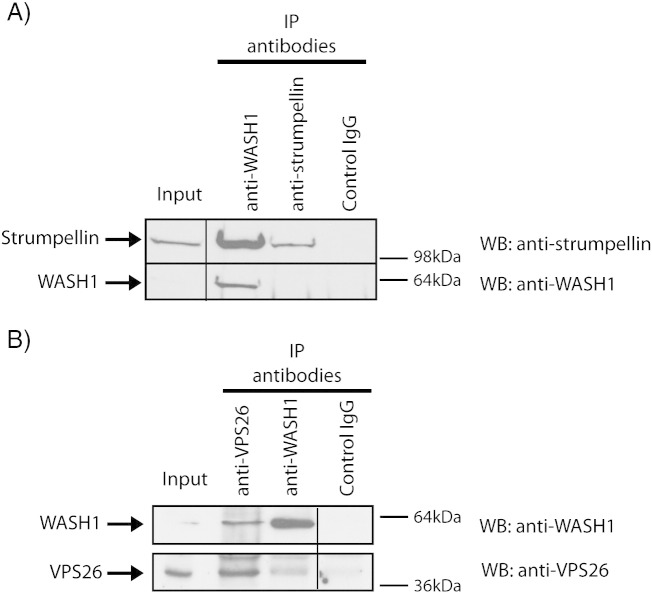
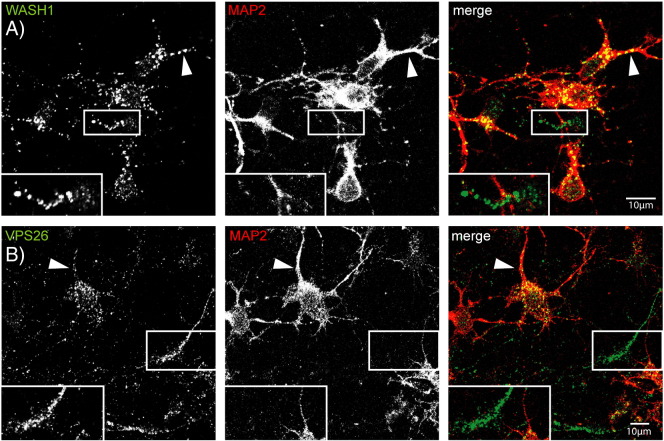
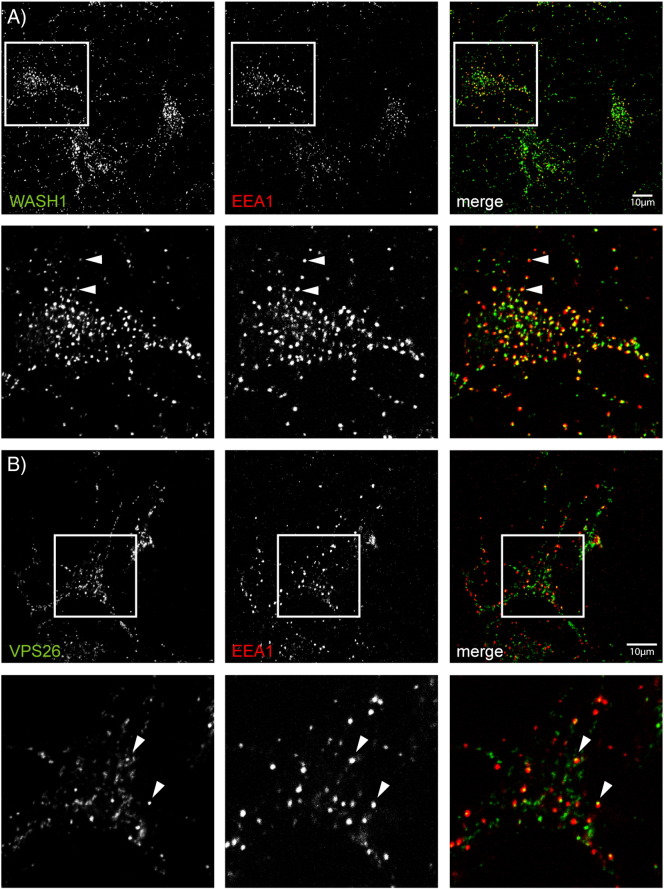
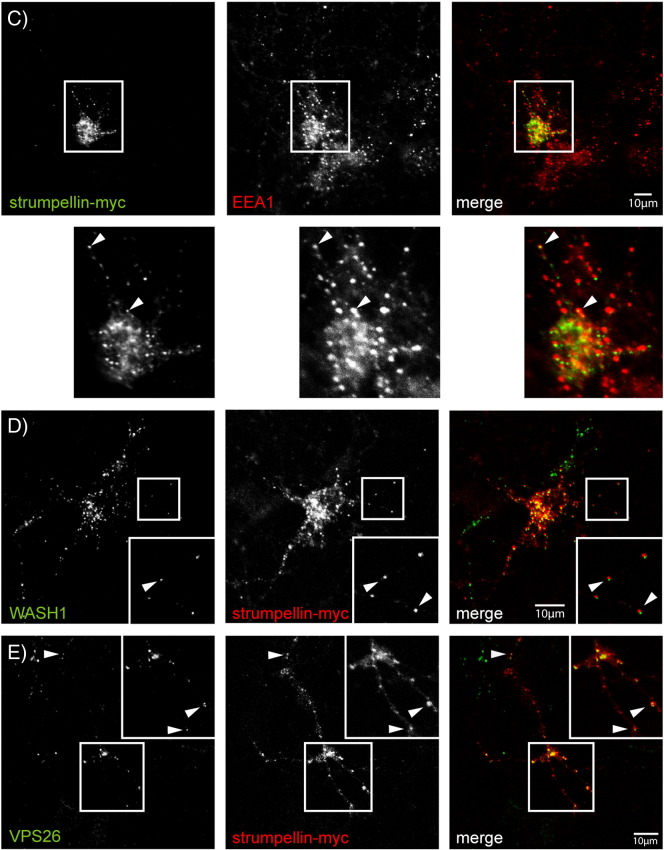
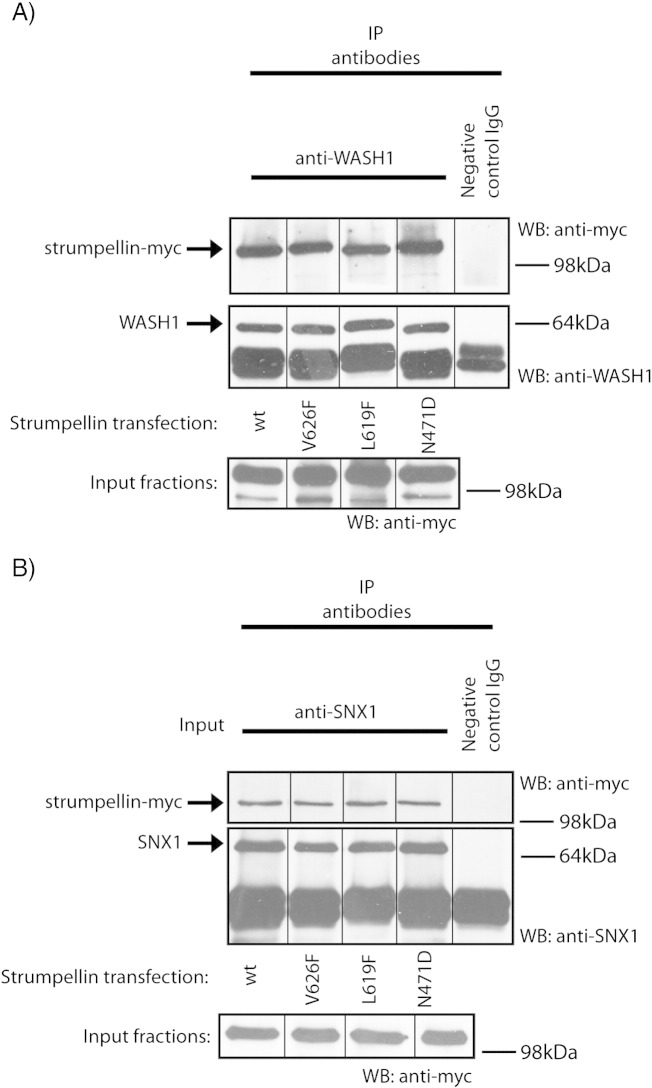
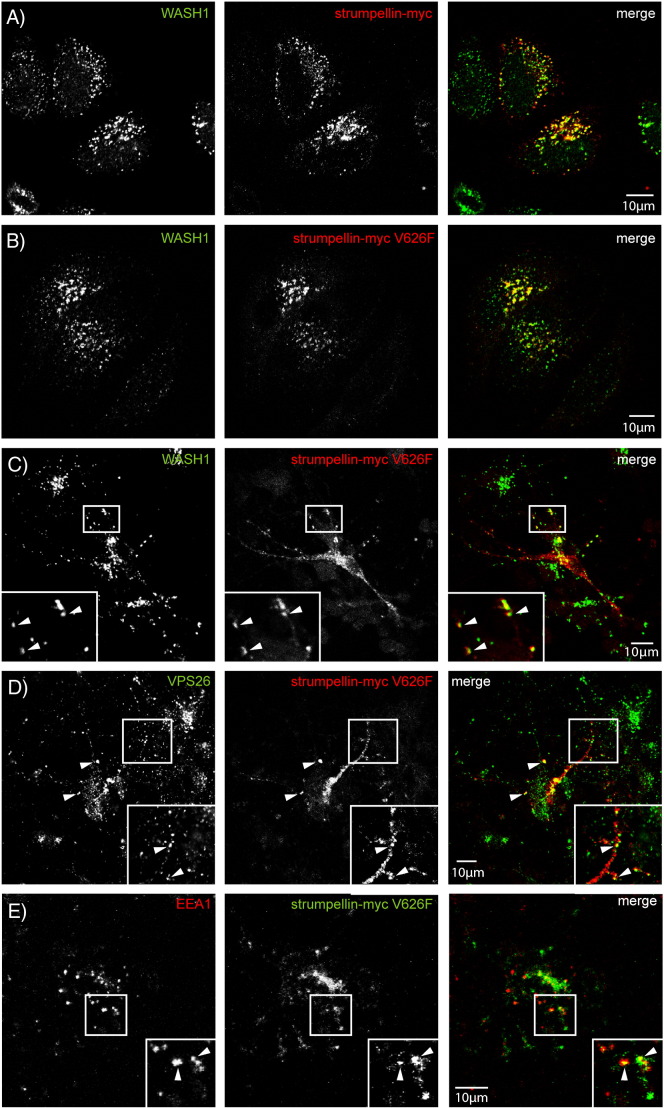
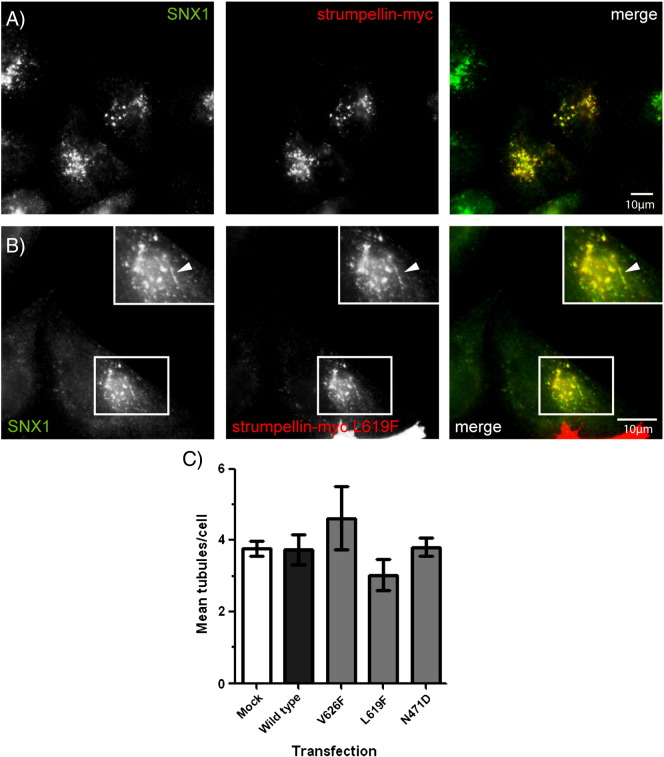
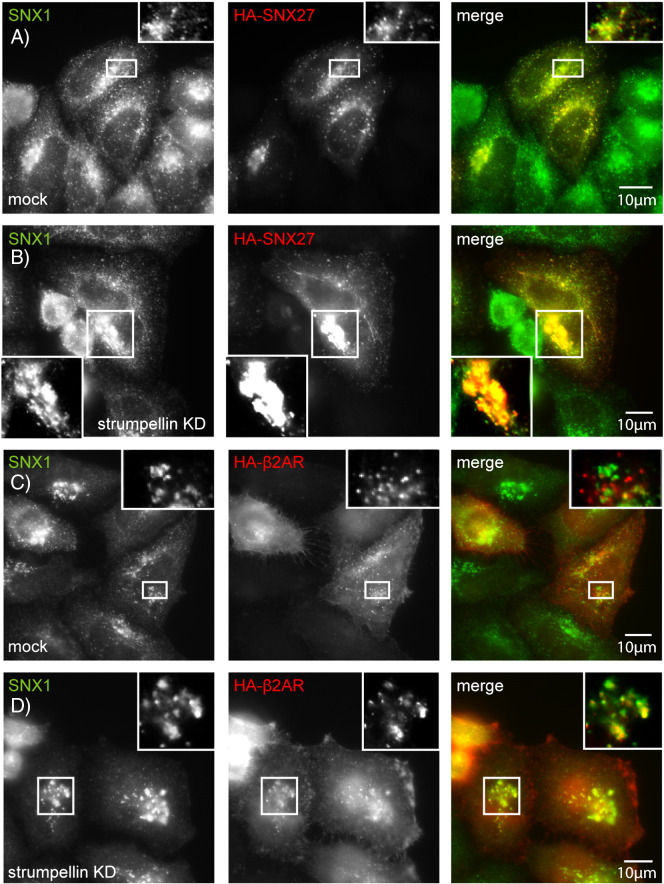
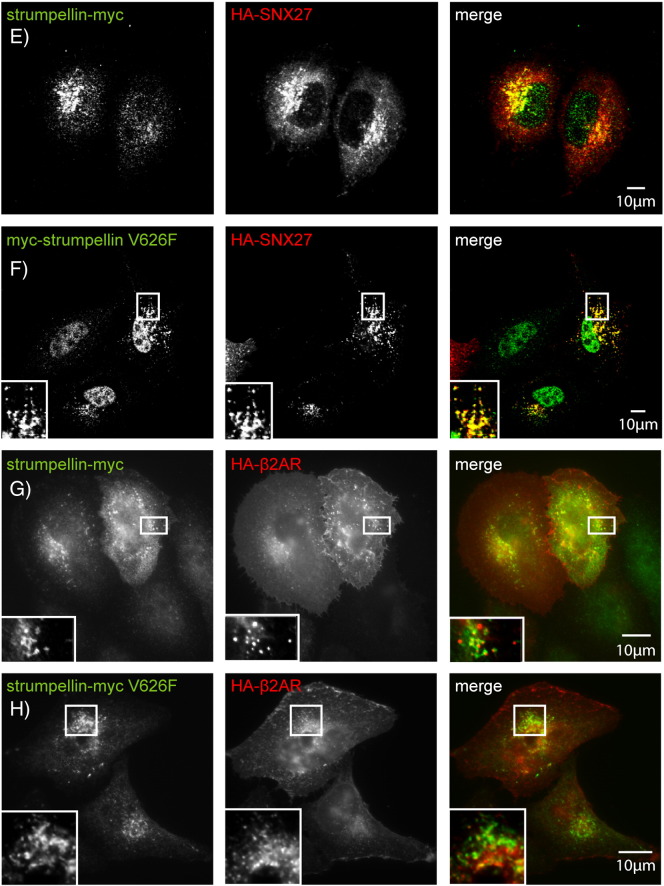
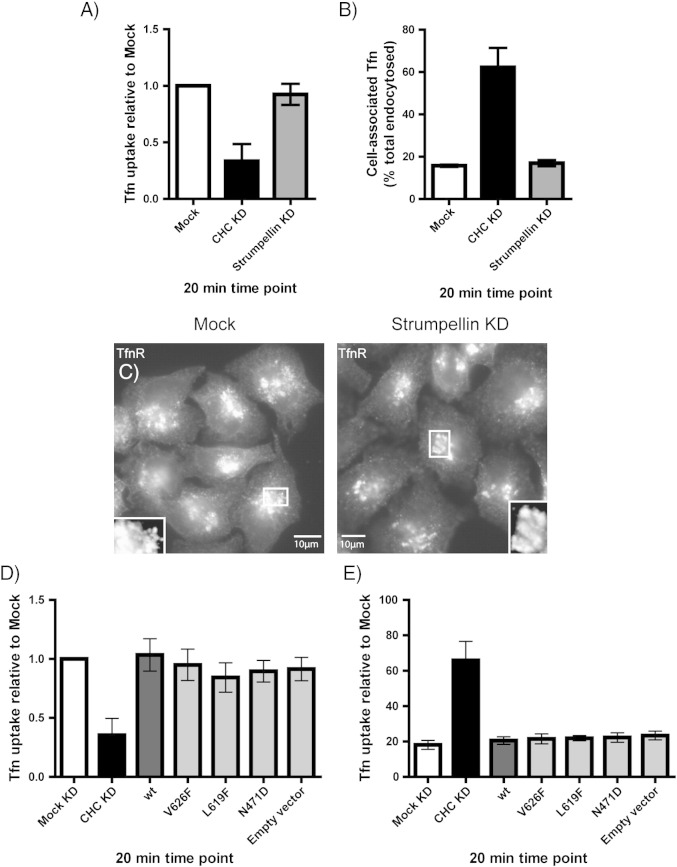
Mutations in the gene encoding strumpellin cause autosomal dominant hereditary spastic paraplegia (HSP), in which there is degeneration of corticospinal tract axons. Strumpellin is a component of the WASH complex, an actin-regulating complex that is recruited to endosomes by interactions with the retromer complex. The WASH complex and its relationship to retromer have not been fully characterised in neurons, and the molecular pathological mechanism of strumpellin mutation is unclear. Here we demonstrate that the WASH complex assembles in the brain, where it interacts with retromer. Members of both complexes co-localise with each other and with endosomes in primary cortical neurons, and are present in somato-dendritic and axonal compartments. We show that strumpellin is not required for normal transferrin receptor traffic, but is required for the correct subcellular distribution of the β-2-adrenergic receptor. However, strumpellin disease mutations do not affect its incorporation into the WASH complex or its subcellular localisation, nor do they have a dominant effect on functions of the WASH complex, including regulation of endosomal tubulation, transferrin receptor traffic or β-2-adrenergic receptor localisation. Models of the WASH complex indicate that it contains a single strumpellin molecule, so in patients with strumpellin mutations, complexes containing wild-type and mutant strumpellin should be present in equal numbers. In most cell types this would provide sufficient functional WASH to allow normal cellular physiology. However, owing to the demands on membrane traffic imposed by their exceptionally long axons, we suggest that corticospinal neurons are especially vulnerable to reductions in functional WASH.
Copyright © 2012 Elsevier B.V. All rights reserved.
Figures
References
-
- Fischer L.R., Culver D.G., Tennant P., Davis A.A., Wang M., Castellano-Sanchez A., Khan J., Polak M.A., Glass J.D. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 2004;185:232–240. - PubMed
-
- Deluca G.C., Ebers G.C., Esiri M.M. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol. Appl. Neurobiol. 2004;30:576–584. - PubMed
-
- Reid E., Rugarli E. In: The Online Metabolic and Molecular Bases of Inherited Diseases. Valle D., Beaudet A., Vogelstein B., Kinzler K., Antonarakis S., Ballabio A., editors. McGraw Hill; New York: 2010.
-
- Salinas S., Proukakis C., Crosby A., Warner T.T. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. - PubMed
-
- Finsterer J., Loscher W., Quasthoff S., Wanschitz J., Auer-Grumbach M., Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012;318:1–18. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
