

Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration
- PMID: 23089226
- PMCID: PMC3568680
- DOI: 10.1016/j.freeradbiomed.2012.10.546
Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration
Abstract
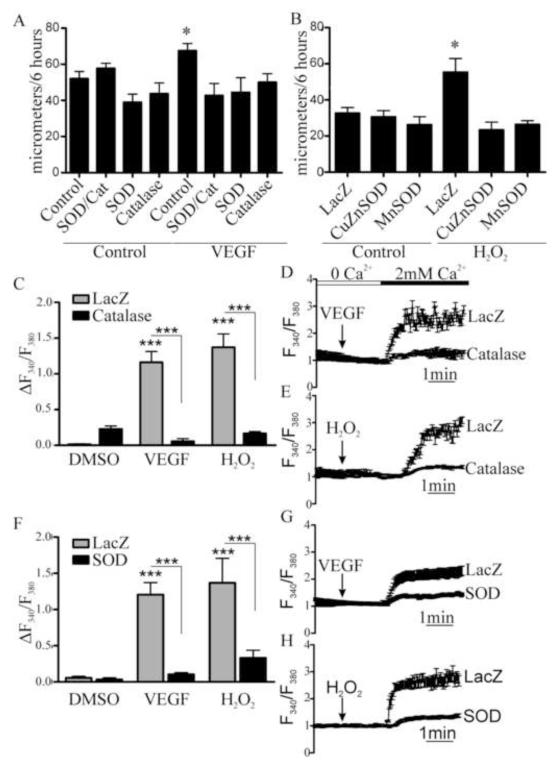
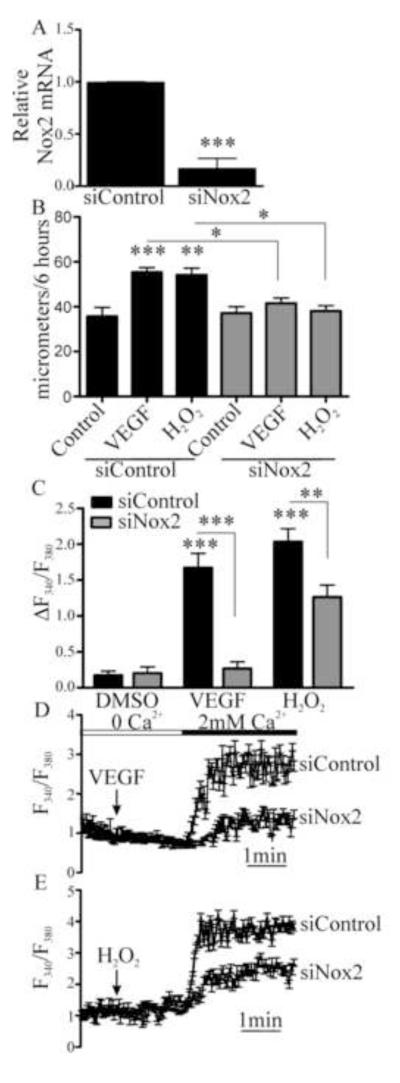
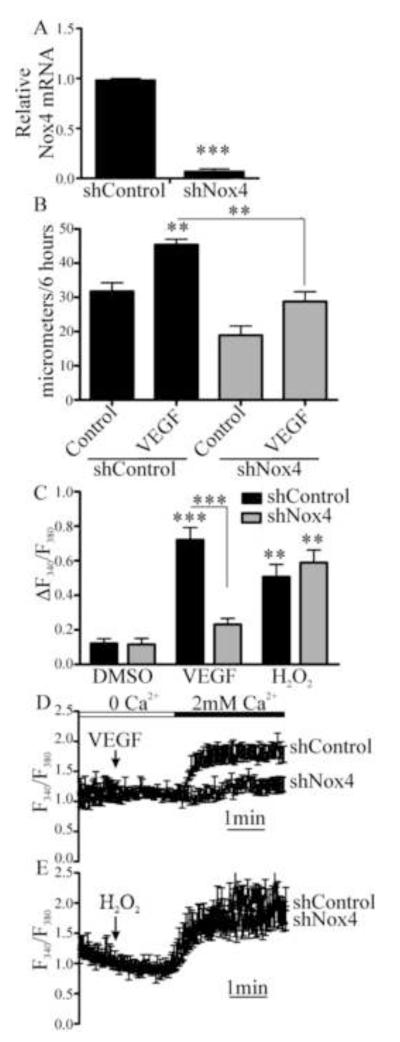
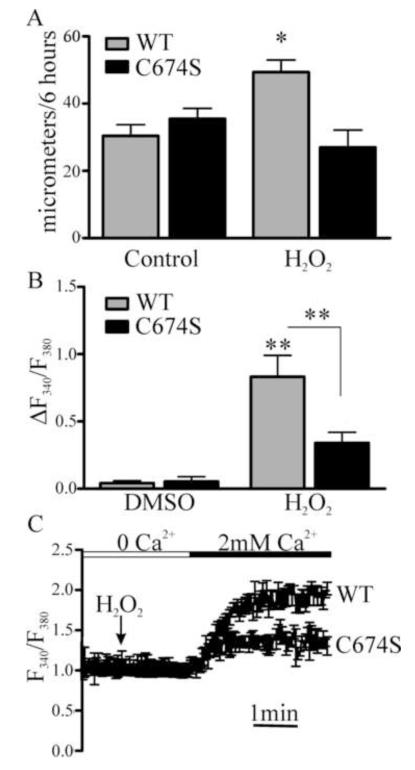
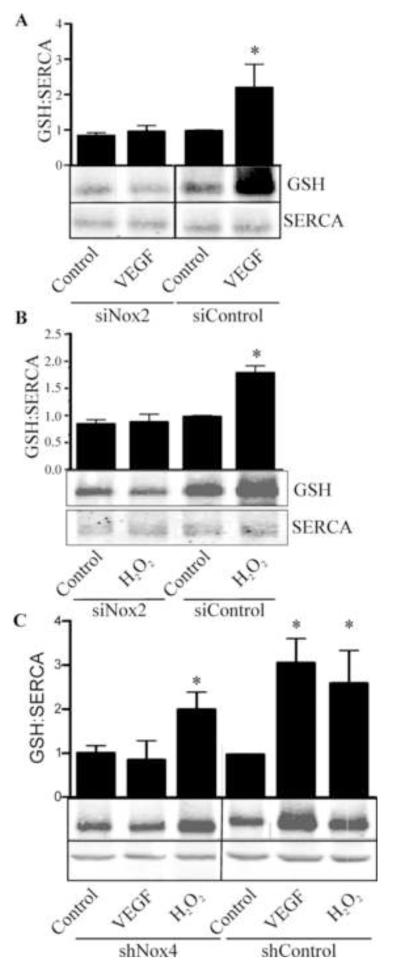
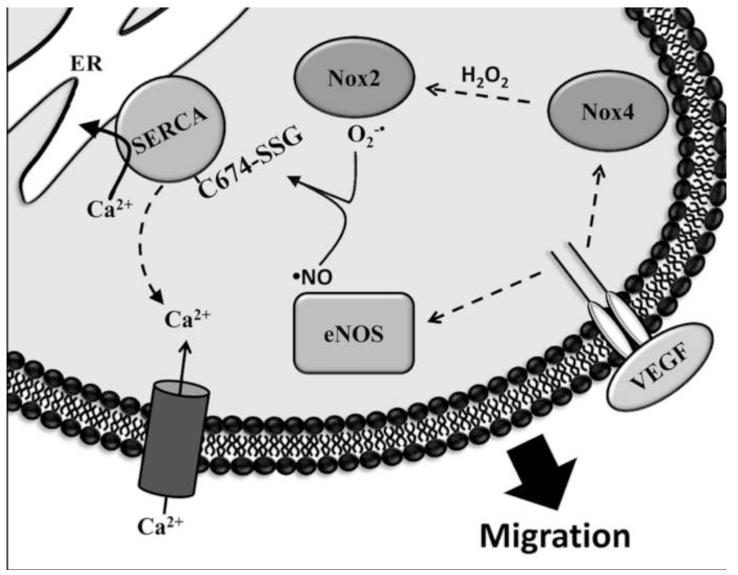
Endothelial cell (EC) migration in response to vascular endothelial growth factor (VEGF) is a critical step in both physiological and pathological angiogenesis. Although VEGF signaling has been extensively studied, the mechanisms by which VEGF-dependent reactive oxygen species (ROS) production affects EC signaling are not well understood. The aim of this study was to elucidate the involvement of Nox2- and Nox4-dependent ROS in VEGF-mediated EC Ca(2+) regulation and migration. VEGF induced migration of human aortic ECs into a scratch wound over 6 h, which was inhibited by overexpression of either catalase or superoxide dismutase (SOD). EC stimulation by micromolar concentrations of H2O2 was inhibited by catalase, but also unexpectedly by SOD. Both VEGF and H2O2 increased S-glutathiolation of SERCA2b and increased Ca(2+) influx into EC, and these events could be blocked by overexpression of catalase or overexpression of SERCA2b in which the reactive cysteine-674 was mutated to a serine. In determining the source of VEGF-mediated ROS production, our studies show that specific knockdown of either Nox2 or Nox4 inhibited VEGF-induced S-glutathiolation of SERCA, Ca(2+) influx, and EC migration. Treatment with H2O2 induced S-glutathiolation of SERCA and EC Ca(2+) influx, overcoming the knockdown of Nox4, but not Nox2, and Amplex red measurements indicated that Nox4 is the source of H2O2. These results demonstrate that VEGF stimulates EC migration through increased S-glutathiolation of SERCA and Ca(2+) influx in a Nox4- and H2O2-dependent manner, requiring Nox2 downstream.
Keywords: Calcium; Endothelial cells; Free radicals; Hydrogen peroxide; Nox2; Nox4; SERCA; VEGF.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
