

Modeling read counts for CNV detection in exome sequencing data
- PMID: 23089826
- PMCID: PMC3517018
- DOI: 10.2202/1544-6115.1732
Modeling read counts for CNV detection in exome sequencing data
Abstract
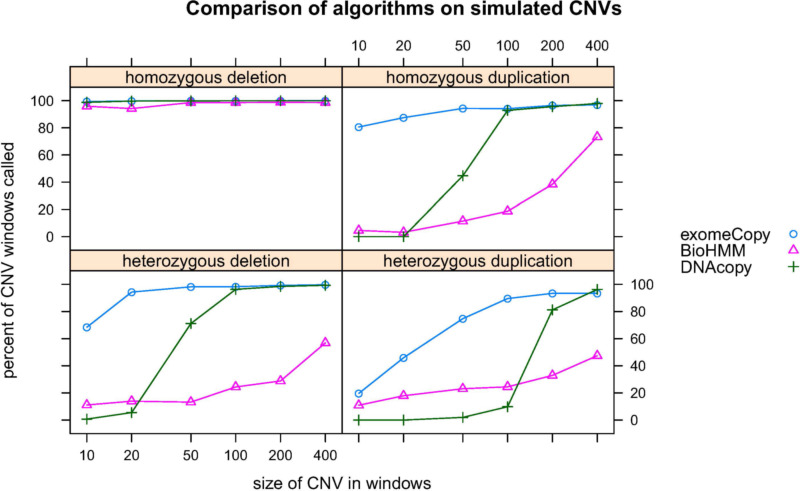
Varying depth of high-throughput sequencing reads along a chromosome makes it possible to observe copy number variants (CNVs) in a sample relative to a reference. In exome and other targeted sequencing projects, technical factors increase variation in read depth while reducing the number of observed locations, adding difficulty to the problem of identifying CNVs. We present a hidden Markov model for detecting CNVs from raw read count data, using background read depth from a control set as well as other positional covariates such as GC-content. The model, exomeCopy, is applied to a large chromosome X exome sequencing project identifying a list of large unique CNVs. CNVs predicted by the model and experimentally validated are then recovered using a cross-platform control set from publicly available exome sequencing data. Simulations show high sensitivity for detecting heterozygous and homozygous CNVs, outperforming normalization and state-of-the-art segmentation methods.
Figures












References
-
- Alkan, C., J. M. Kidd, T. Marques-Bonet, G. Aksay, F. Antonacci, F. Hormozdiari, J. O. Kitzman, C. Baker, M. Malig, O. Mutlu, S. C. Sahinalp, R. A. Gibbs, and E. E. Eichler (2009): “Personalized copy number and segmental duplication maps using next-generation sequencing,” Nature Genetics, 41, 1061–1067. - PMC - PubMed
-
- Benjamini, Y. and T. P. Speed (2011): “Estimation and correction for GC-content bias in high throughput sequencing,” Technical report, University of California at Berkeley.
-
- Bliss, C. I. and R. A. Fisher (1953): “Fitting the Negative Binomial Distribution to Biological Data,” Biometrics, 9.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous