

Alteration of the oxygen-dependent reactivity of de novo Due Ferri proteins
- PMID: 23089864
- PMCID: PMC3568993
- DOI: 10.1038/nchem.1454
Alteration of the oxygen-dependent reactivity of de novo Due Ferri proteins
Erratum in
- Nat Chem. 2012 Dec;4(12):1050. Szyperski, Thomas G [corrected to Szyperski, Thomas]
Abstract
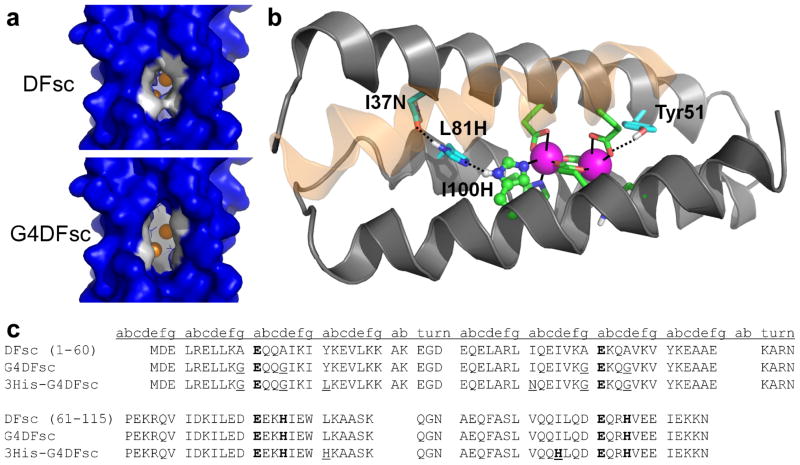
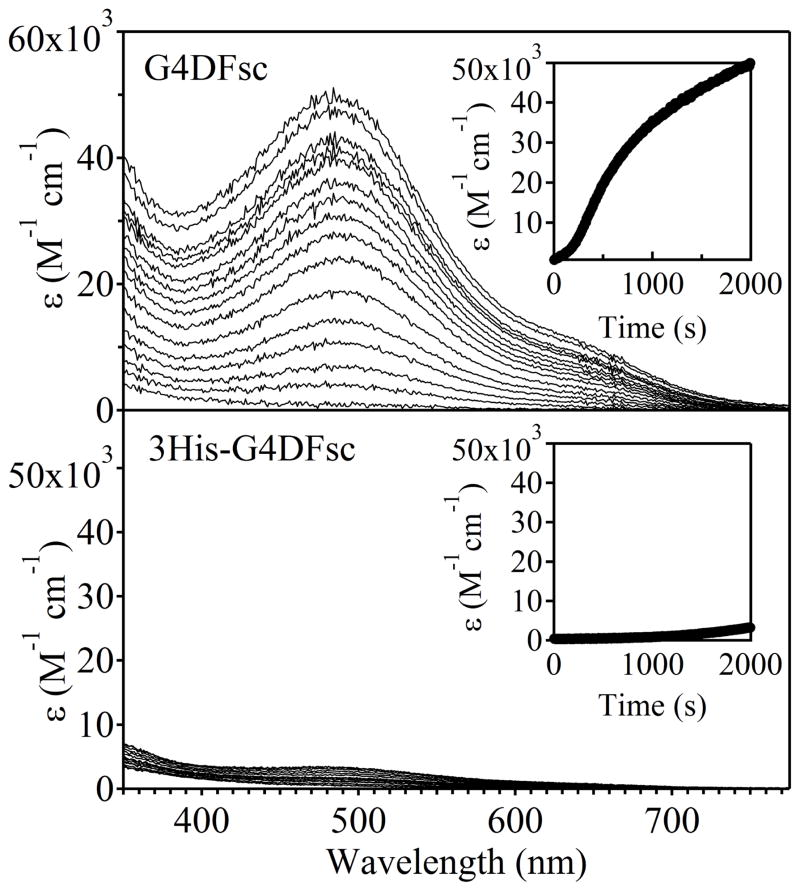
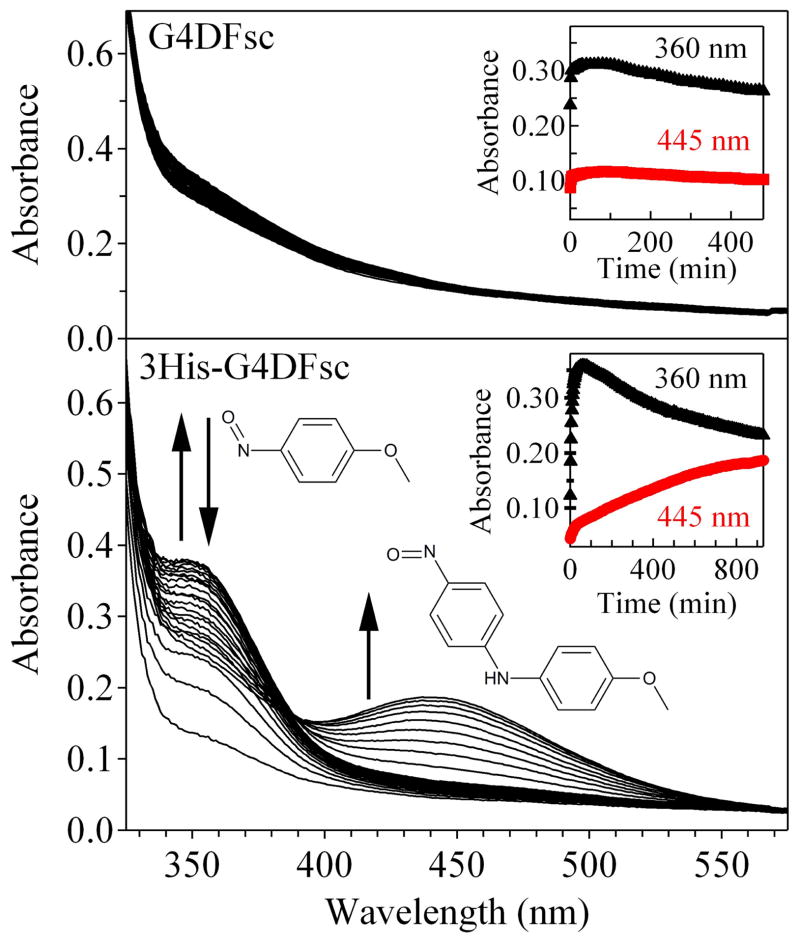
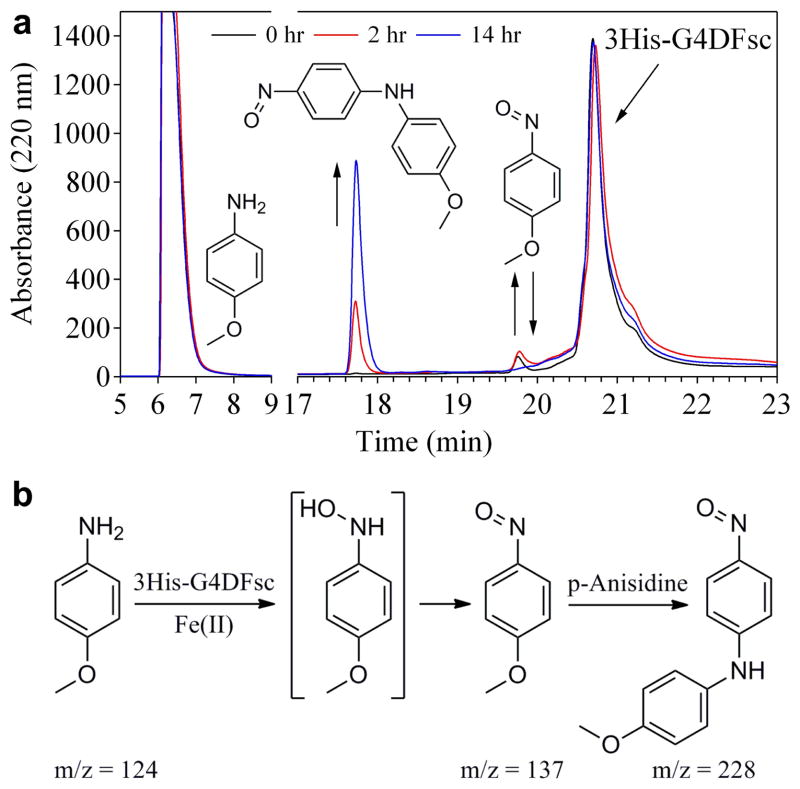
De novo proteins provide a unique opportunity to investigate the structure-function relationships of metalloproteins in a minimal, well-defined and controlled scaffold. Here, we describe the rational programming of function in a de novo designed di-iron carboxylate protein from the Due Ferri family. Originally created to catalyse the O(2)-dependent, two-electron oxidation of hydroquinones, the protein was reprogrammed to catalyse the selective N-hydroxylation of arylamines by remodelling the substrate access cavity and introducing a critical third His ligand to the metal-binding cavity. Additional second- and third-shell modifications were required to stabilize the His ligand in the core of the protein. These structural changes resulted in at least a 10(6)-fold increase in the relative rate between the arylamine N-hydroxylation and hydroquinone oxidation reactions. This result highlights the potential for using de novo proteins as scaffolds for future investigations of the geometric and electronic factors that influence the catalytic tuning of di-iron active sites.
Figures


Comment in
-
Protein design: Engineering di-iron enzymes.Nat Chem. 2012 Nov;4(11):868-9. doi: 10.1038/nchem.1483. Nat Chem. 2012. PMID: 23089858 No abstract available.
Similar articles
-
De Novo Design of Four-Helix Bundle Metalloproteins: One Scaffold, Diverse Reactivities.Acc Chem Res. 2019 May 21;52(5):1148-1159. doi: 10.1021/acs.accounts.8b00674. Epub 2019 Apr 11. Acc Chem Res. 2019. PMID: 30973707 Free PMC article.
-
Ferrous binding to the multicopper oxidases Saccharomyces cerevisiae Fet3p and human ceruloplasmin: contributions to ferroxidase activity.J Am Chem Soc. 2004 Jun 2;126(21):6579-89. doi: 10.1021/ja049220t. J Am Chem Soc. 2004. PMID: 15161286
-
Crystal structure of PnpCD, a two-subunit hydroquinone 1,2-dioxygenase, reveals a novel structural class of Fe2+-dependent dioxygenases.J Biol Chem. 2015 Oct 2;290(40):24547-60. doi: 10.1074/jbc.M115.673558. Epub 2015 Aug 24. J Biol Chem. 2015. PMID: 26304122 Free PMC article.
-
Structural principles for computational and de novo design of 4Fe-4S metalloproteins.Biochim Biophys Acta. 2016 May;1857(5):531-538. doi: 10.1016/j.bbabio.2015.10.001. Epub 2015 Oct 9. Biochim Biophys Acta. 2016. PMID: 26449207 Free PMC article. Review.
-
Tertiary templates for the design of diiron proteins.Curr Opin Struct Biol. 1999 Aug;9(4):500-8. doi: 10.1016/S0959-440X(99)80071-2. Curr Opin Struct Biol. 1999. PMID: 10449377 Review.
Cited by
-
De Novo Design of a Self-Assembled Artificial Copper Peptide that Activates and Reduces Peroxide.ACS Catal. 2021 Aug 20;11(16):10267-10278. doi: 10.1021/acscatal.1c02132. Epub 2021 Aug 3. ACS Catal. 2021. PMID: 36188417 Free PMC article.
-
Molecular-Level Insight into the Differential Oxidase and Oxygenase Reactivities of de Novo Due Ferri Proteins.J Am Chem Soc. 2015 Jul 29;137(29):9302-14. doi: 10.1021/jacs.5b03524. Epub 2015 Jul 15. J Am Chem Soc. 2015. PMID: 26090726 Free PMC article.
-
An efficient, step-economical strategy for the design of functional metalloproteins.Nat Chem. 2019 May;11(5):434-441. doi: 10.1038/s41557-019-0218-9. Epub 2019 Feb 18. Nat Chem. 2019. PMID: 30778140 Free PMC article.
-
A de novo designed metalloenzyme for the hydration of CO2.Angew Chem Int Ed Engl. 2014 Jul 21;53(30):7900-3. doi: 10.1002/anie.201404925. Epub 2014 Jun 18. Angew Chem Int Ed Engl. 2014. PMID: 24943466 Free PMC article.
-
Designed multi-stranded heme binding β-sheet peptides in membrane.Chem Sci. 2016 Apr 21;7(4):2563-2571. doi: 10.1039/c5sc04108b. Epub 2016 Dec 17. Chem Sci. 2016. PMID: 28660027 Free PMC article.
References
-
- Kolberg M, Strand KR, Graff P, Andersson KK. Structure, function, and mechanism of ribonucleotide reductases. BBA-Proteins Proteom. 2004;1699:1–34. - PubMed
-
- Berthold DA, Stenmark P. Membrane-bound diiron carboxylate proteins. Annu Rev Plant Biol. 2003;54:497–517. - PubMed
-
- Solomon EI, et al. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem Rev. 2000;100:235–350. - PubMed
-
- Lippard SJ. Hydroxylation of C–H bonds at carboxylate-bridged diiron centres. Philos T R Soc A. 2005;363:861–877. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
