

ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions
- PMID: 23090257
- PMCID: PMC3591838
- DOI: 10.1038/nrg3306
ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions
Abstract
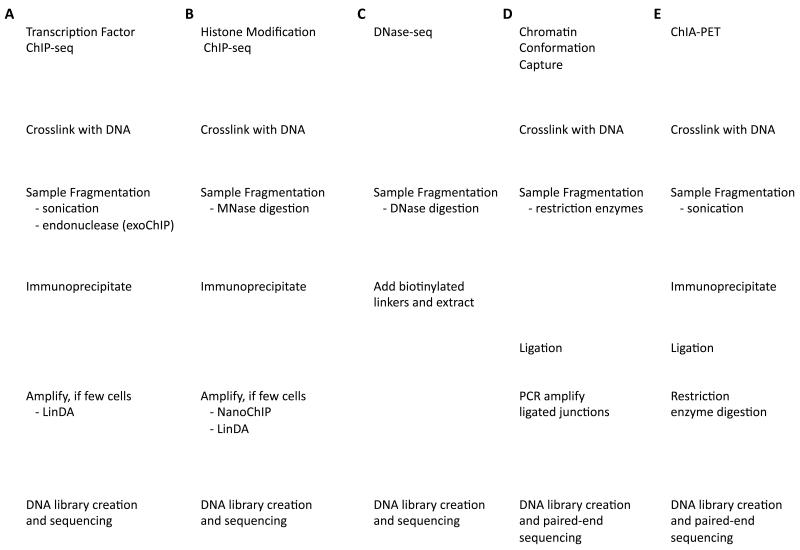
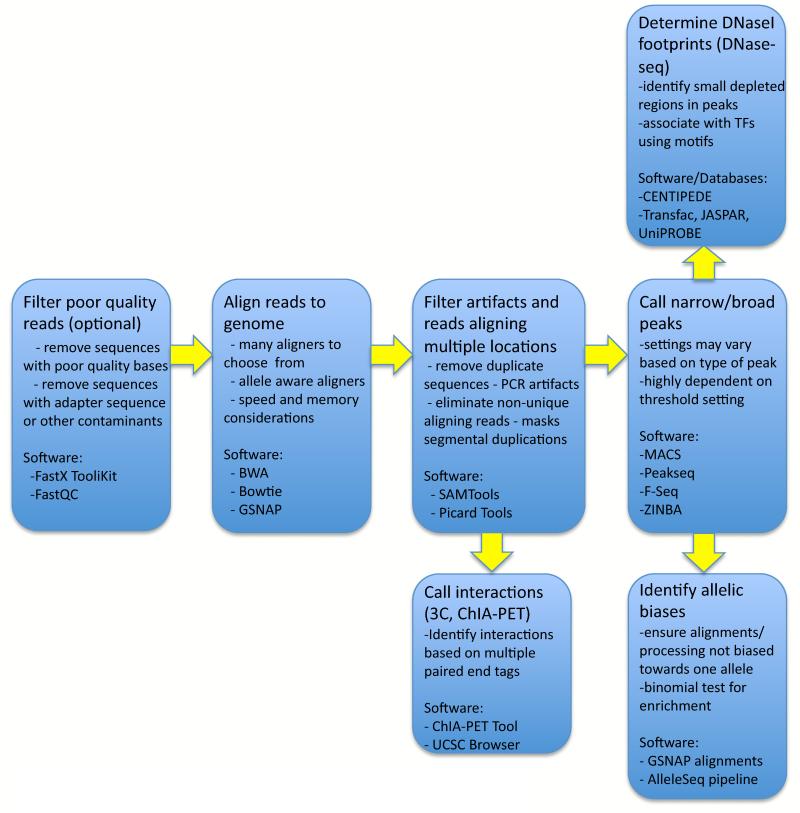
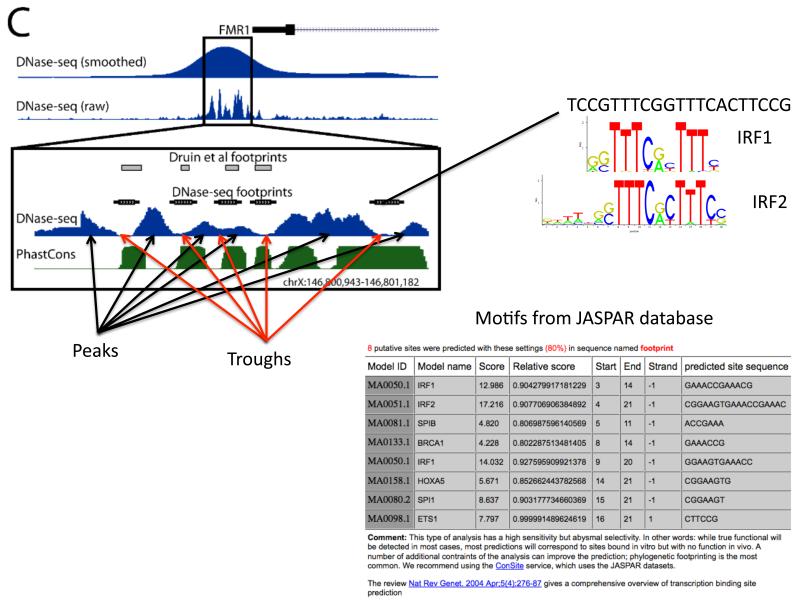
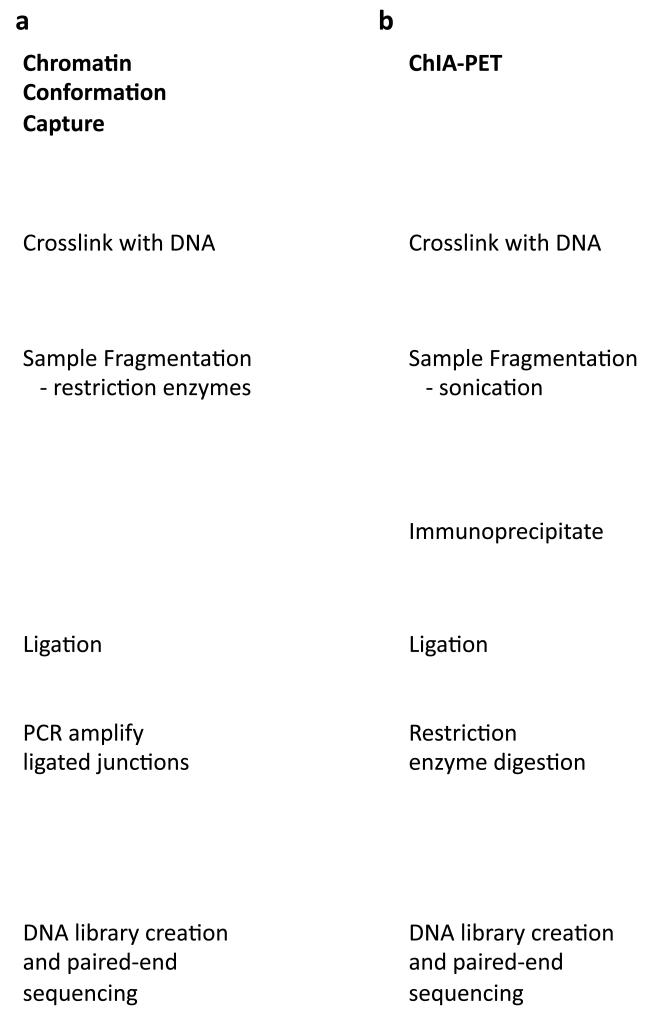
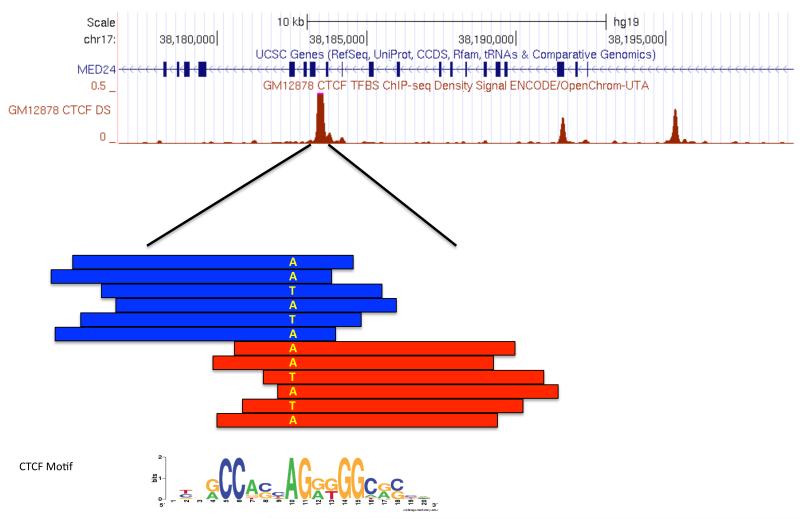
Chromatin immunoprecipitation experiments followed by sequencing (ChIP-seq) detect protein-DNA binding events and chemical modifications of histone proteins. Challenges in the standard ChIP-seq protocol have motivated recent enhancements in this approach, such as reducing the number of cells that are required and increasing the resolution. Complementary experimental approaches - for example, DNaseI hypersensitive site mapping and analysis of chromatin interactions that are mediated by particular proteins - provide additional information about DNA-binding proteins and their function. These data are now being used to identify variability in the functions of DNA-binding proteins across genomes and individuals. In this Review, I describe the latest advances in methods to detect and functionally characterize DNA-bound proteins.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
