

Accurate simulation and detection of coevolution signals in multiple sequence alignments
- PMID: 23091608
- PMCID: PMC3473043
- DOI: 10.1371/journal.pone.0047108
Accurate simulation and detection of coevolution signals in multiple sequence alignments
Abstract
Background: While the conserved positions of a multiple sequence alignment (MSA) are clearly of interest, non-conserved positions can also be important because, for example, destabilizing effects at one position can be compensated by stabilizing effects at another position. Different methods have been developed to recognize the evolutionary relationship between amino acid sites, and to disentangle functional/structural dependencies from historical/phylogenetic ones.
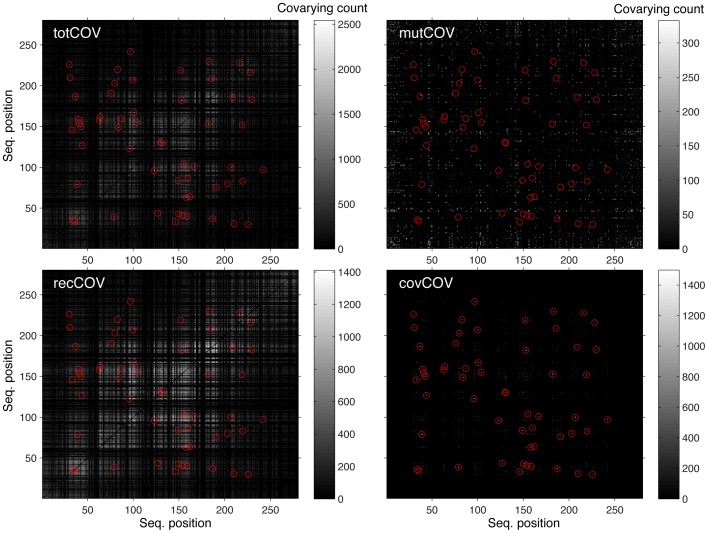
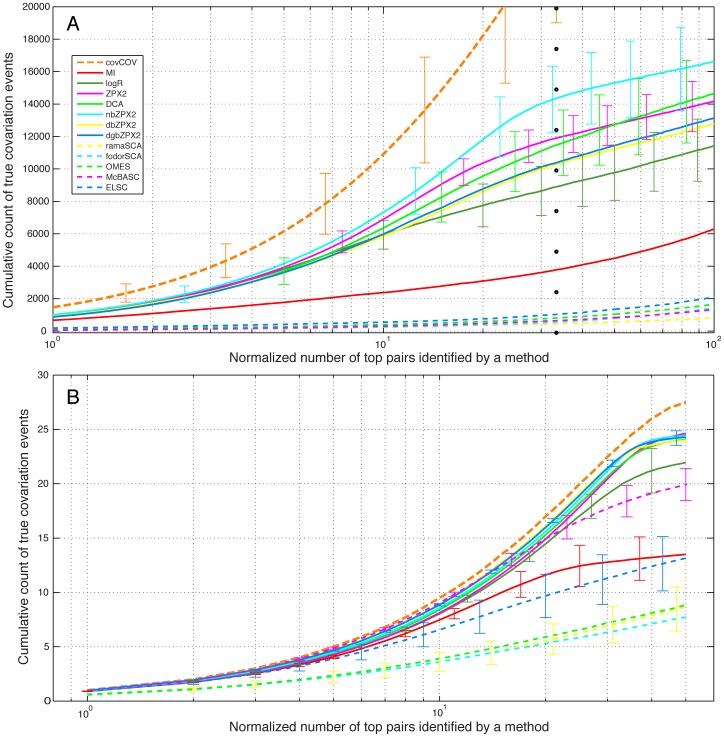
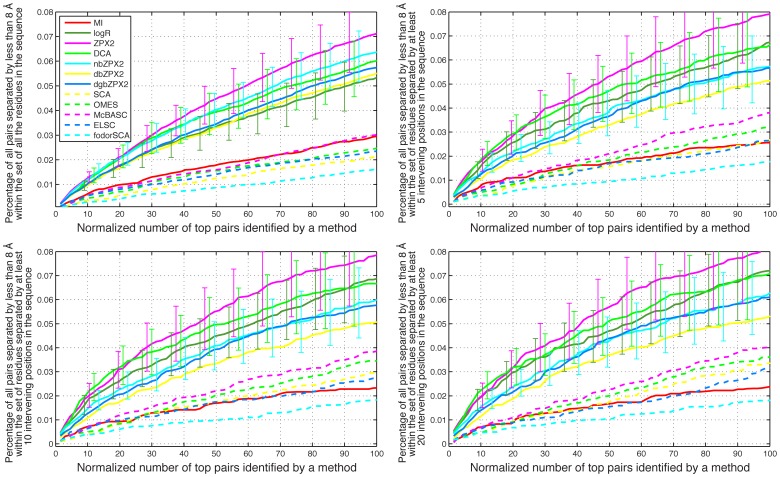
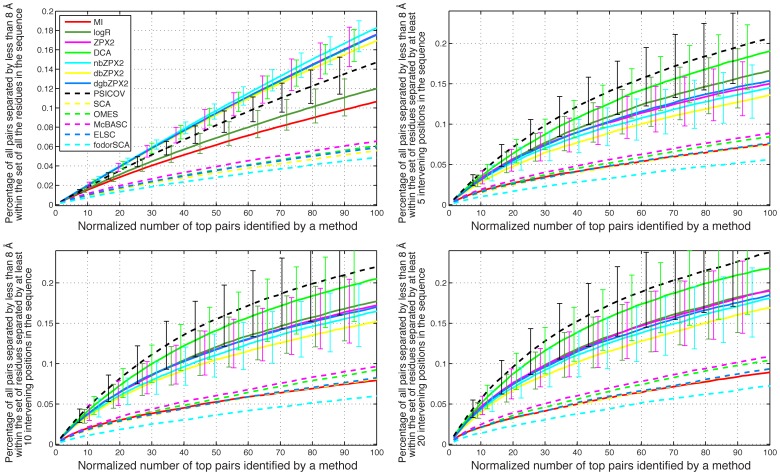
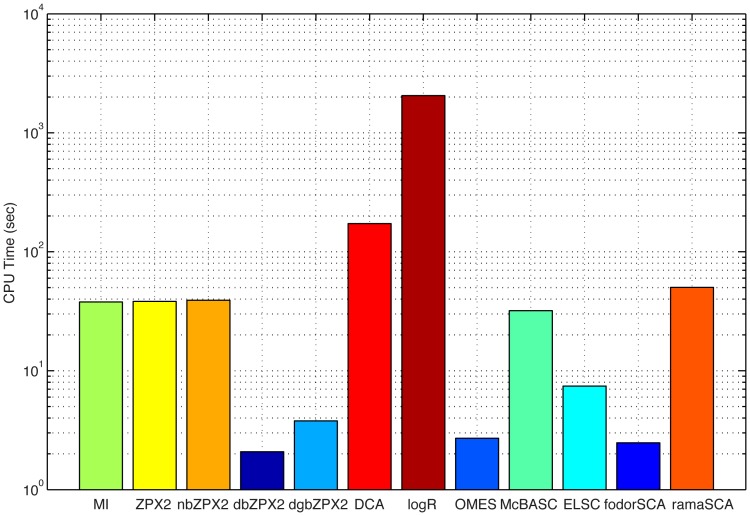
Methodology/principal findings: We have used two complementary approaches to test the efficacy of these methods. In the first approach, we have used a new program, MSAvolve, for the in silico evolution of MSAs, which records a detailed history of all covarying positions, and builds a global coevolution matrix as the accumulated sum of individual matrices for the positions forced to co-vary, the recombinant coevolution, and the stochastic coevolution. We have simulated over 1600 MSAs for 8 protein families, which reflect sequences of different sizes and proteins with widely different functions. The calculated coevolution matrices were compared with the coevolution matrices obtained for the same evolved MSAs with different coevolution detection methods. In a second approach we have evaluated the capacity of the different methods to predict close contacts in the representative X-ray structures of an additional 150 protein families using only experimental MSAs.
Conclusions/significance: Methods based on the identification of global correlations between pairs were found to be generally superior to methods based only on local correlations in their capacity to identify coevolving residues using either simulated or experimental MSAs. However, the significant variability in the performance of different methods with different proteins suggests that the simulation of MSAs that replicate the statistical properties of the experimental MSA can be a valuable tool to identify the coevolution detection method that is most effective in each case.
Conflict of interest statement
Figures


 .
.
References
-
- Atchley WR, Wollenberg KR, Fitch WM, Terhalle W, Dress AW (2000) Correlations among amino acid sites in bHLH protein domains: an information theoretic analysis. Mol Biol Evol 17: 164–178. - PubMed
-
- Horner DS, Pirovano W, Pesole G (2008) Correlated substitution analysis and the prediction of amino acid structural contacts. Brief Bioinform 9: 46–56. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
