

Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism
- PMID: 23093779
- PMCID: PMC3484445
- DOI: 10.1172/JCI63377
Osteoblasts mediate the adverse effects of glucocorticoids on fuel metabolism
Abstract
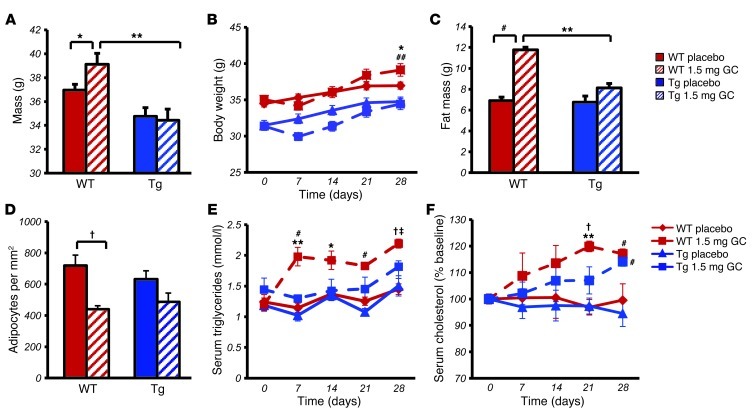
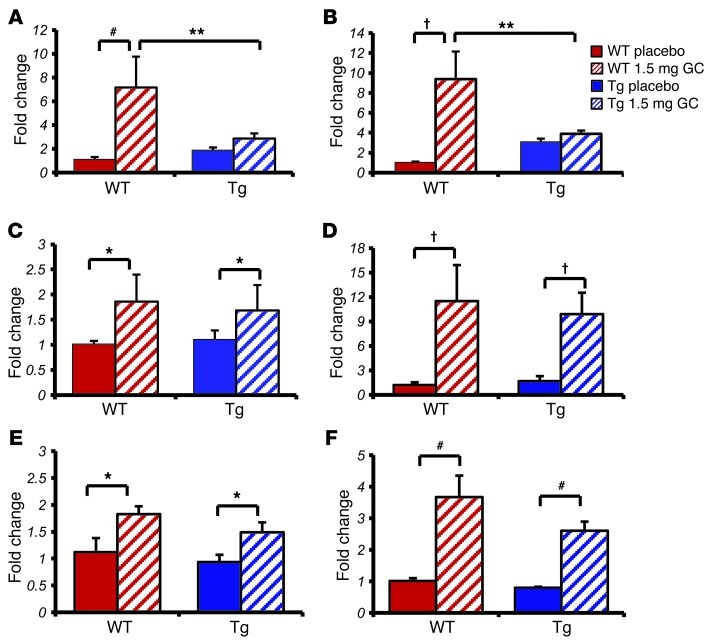
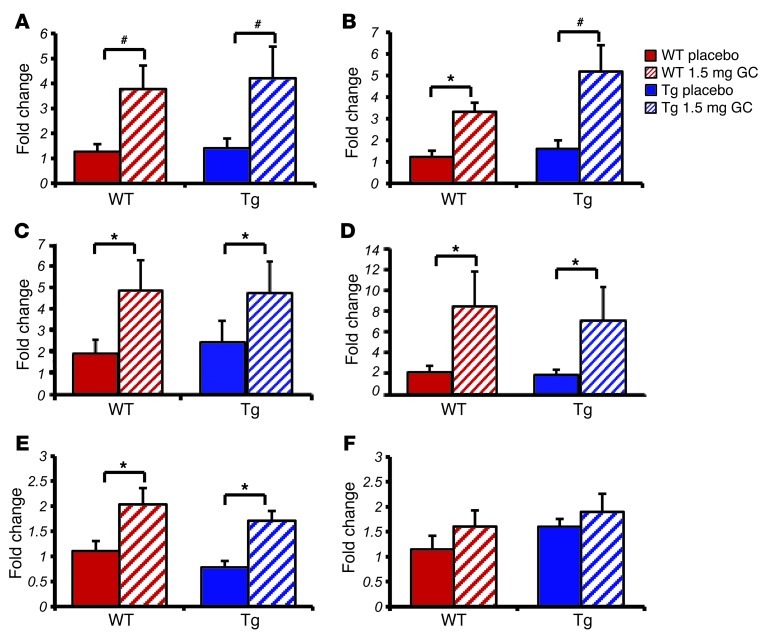
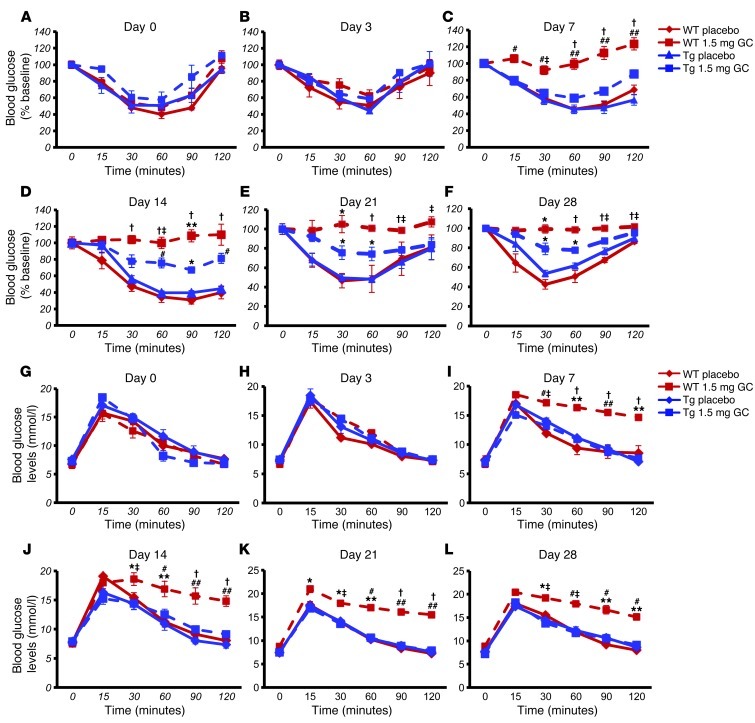
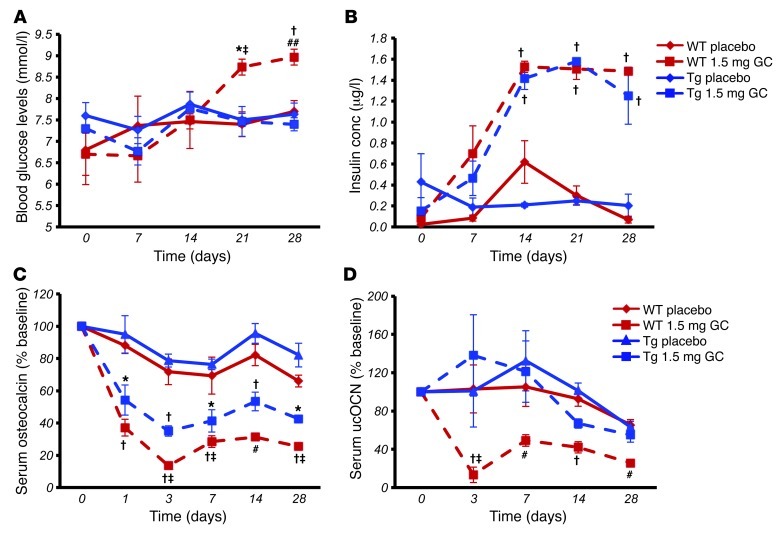
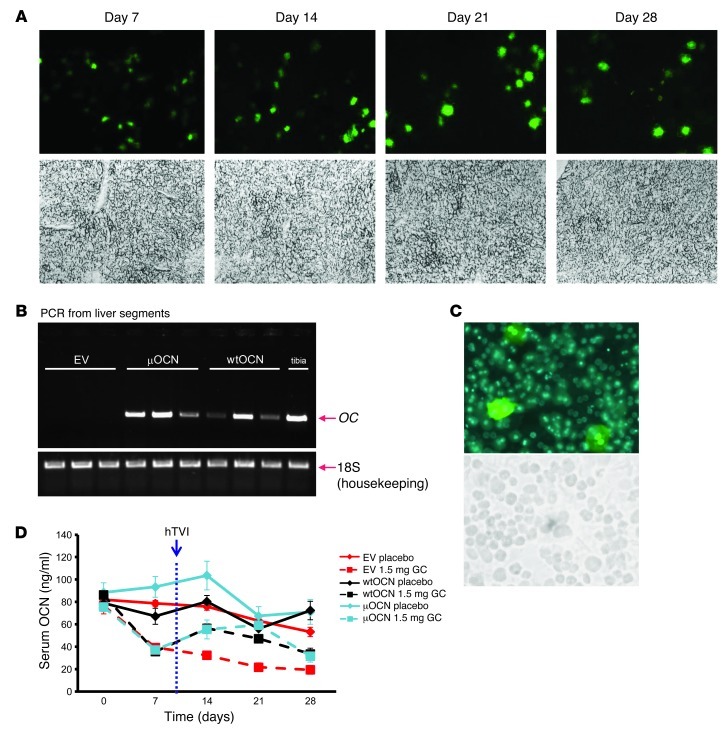
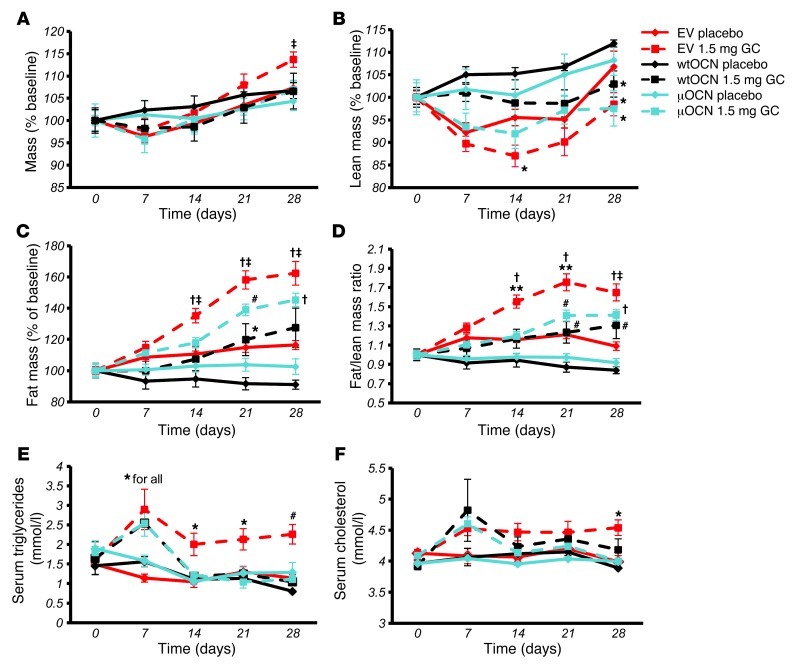
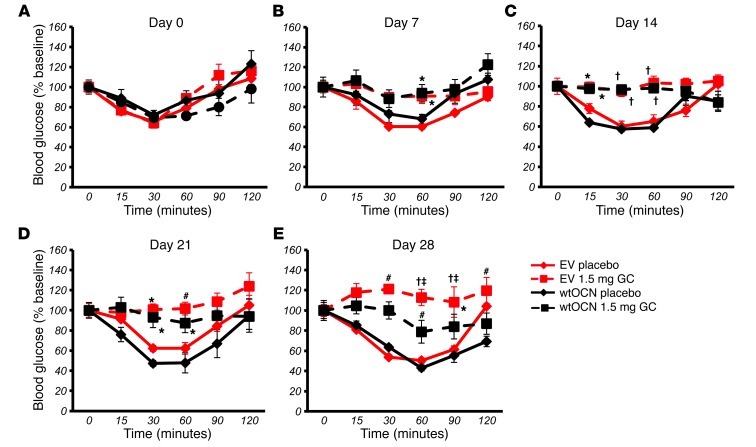
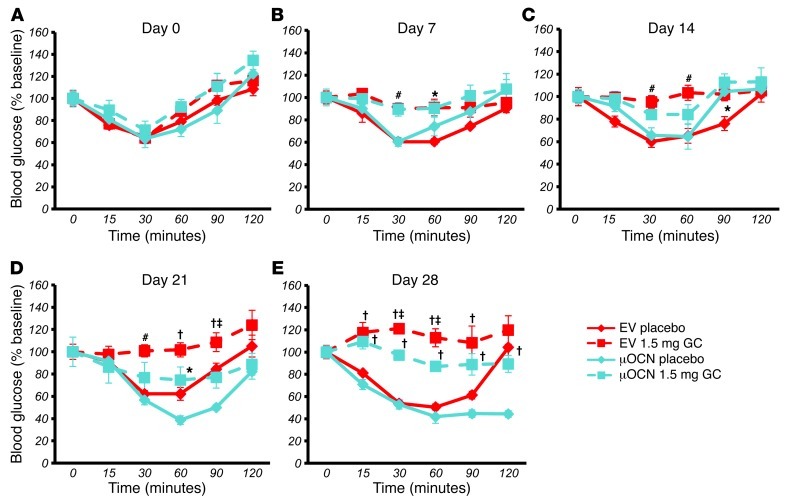
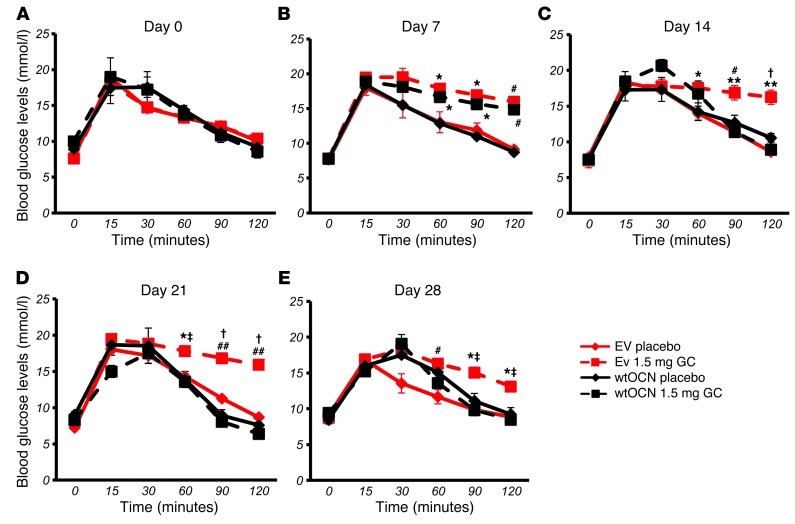
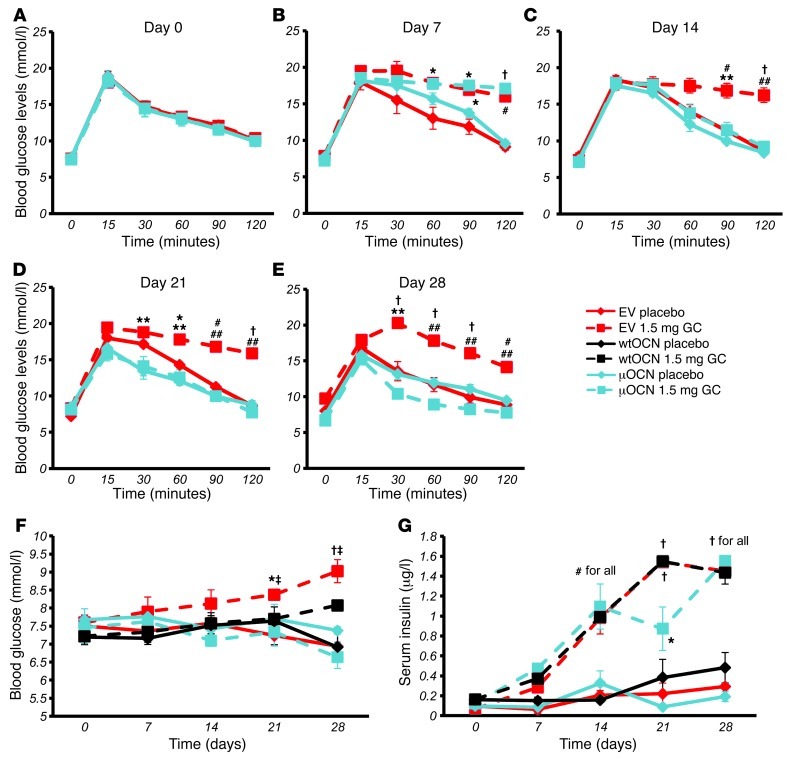
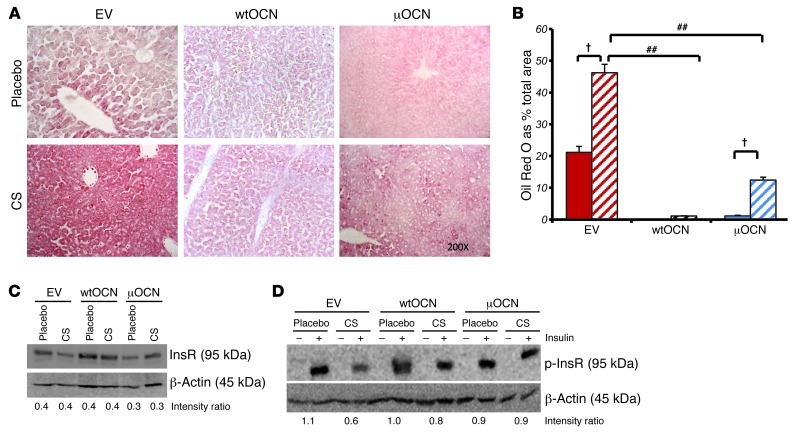
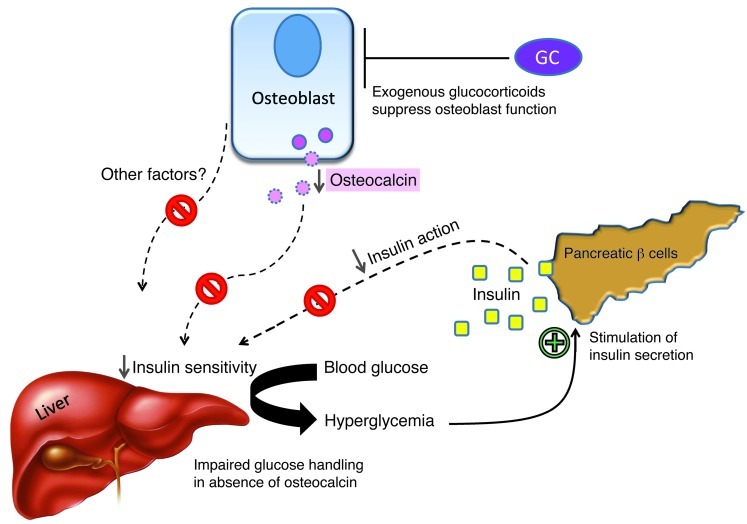
Long-term glucocorticoid treatment is associated with numerous adverse outcomes, including weight gain, insulin resistance, and diabetes; however, the pathogenesis of these side effects remains obscure. Glucocorticoids also suppress osteoblast function, including osteocalcin synthesis. Osteocalcin is an osteoblast-specific peptide that is reported to be involved in normal murine fuel metabolism. We now demonstrate that osteoblasts play a pivotal role in the pathogenesis of glucocorticoid-induced dysmetabolism. Osteoblast-targeted disruption of glucocorticoid signaling significantly attenuated the suppression of osteocalcin synthesis and prevented the development of insulin resistance, glucose intolerance, and abnormal weight gain in corticosterone-treated mice. Nearly identical effects were observed in glucocorticoid-treated animals following heterotopic (hepatic) expression of both carboxylated and uncarboxylated osteocalcin through gene therapy, which additionally led to a reduction in hepatic lipid deposition and improved phosphorylation of the insulin receptor. These data suggest that the effects of exogenous high-dose glucocorticoids on insulin target tissues and systemic energy metabolism are mediated, at least in part, through the skeleton.
Figures
Comment in
-
New mechanisms of glucocorticoid-induced insulin resistance: make no bones about it.J Clin Invest. 2012 Nov;122(11):3854-7. doi: 10.1172/JCI66180. Epub 2012 Oct 24. J Clin Invest. 2012. PMID: 23093783 Free PMC article.
-
Bone: Bare bones of glucocorticoid effects on metabolism.Nat Rev Rheumatol. 2012 Dec;8(12):694. doi: 10.1038/nrrheum.2012.203. Epub 2012 Nov 13. Nat Rev Rheumatol. 2012. PMID: 23147889 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
