

Prader-Willi syndrome: current understanding of cause and diagnosis
- PMID: 2309779
- PMCID: PMC5493042
- DOI: 10.1002/ajmg.1320350306
Prader-Willi syndrome: current understanding of cause and diagnosis
Abstract
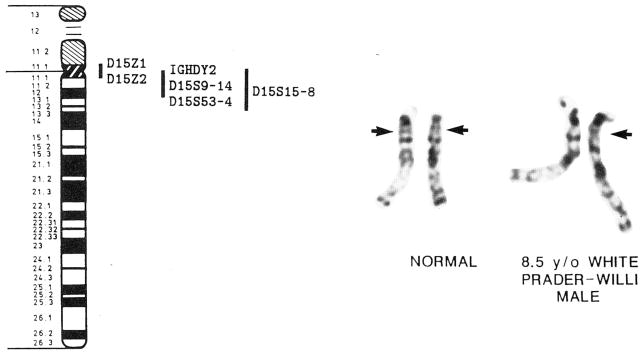
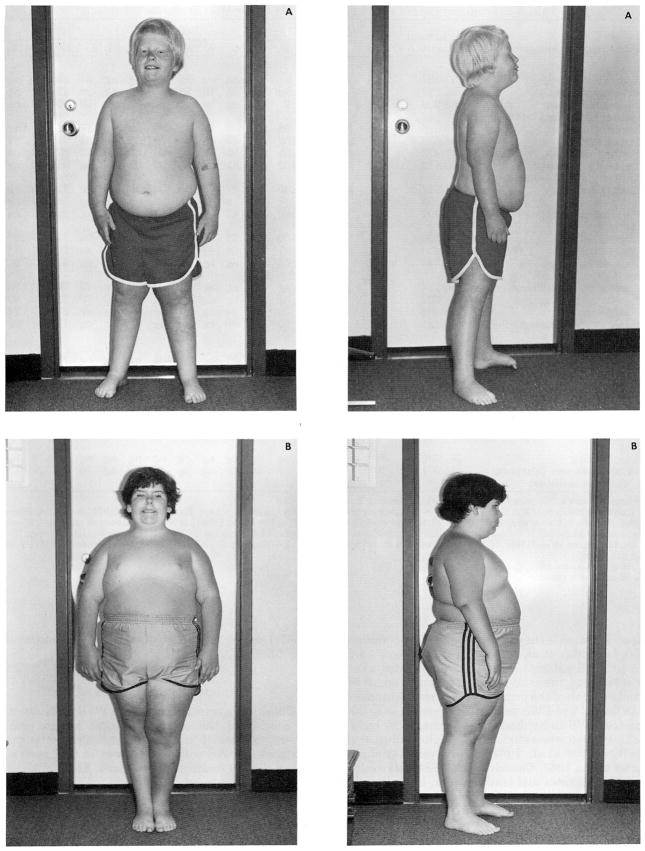
Prader-Willi syndrome (PWS) is characterized by hypotonia, obesity, hypogonadism, short stature, small hands and feet, mental deficiency, a characteristic face, and an interstitial deletion of the proximal long arm of chromosome 15 in about one-half of the patients. The incidence is estimated to be about 1 in 25,000, and PWS is the most common syndromal cause of human obesity. DNA abnormalities, usually deletions or duplications of chromosome 15, have been identified in individuals with PWS with or without recognizable chromosome 15 deletions. Paternal origin of the chromosome 15 deletion by cytogenetic and DNA studies has been found in nearly all PWS individuals studied. No cytogenetic evidence for chromosome breakage has been identified, although an environmental cause (e.g., paternal hydrocarbon-exposed occupations) of the chromosome 15 abnormality has been proposed. PWS patients with the chromosome 15 deletion are more prone to hypopigmentation compared with PWS individuals with normal chromosomes, but no other clinical differences are consistently identified between those with and without the chromosome deletion. Anthropometric, dermatoglyphic, and other clinical findings indicate homogeneity of PWS patients with the chromosome deletion and heterogeneity of the nondeletion patients. A review of our current understanding of the major clinical, cytogenetic, and DNA findings is presented, and clinical manifestations and cytogenetic abnormalities are summarized from the literature.
Figures
References
-
- Afifi AK, Zellweger H. Pathology of muscular hypotonia in the Prader-Willi syndrome. J Neurol Sci. 1969;9:49–61. - PubMed
-
- Aurias A, Prieur M, Dutrillaux B, Lejeune J. Systemic analysis of 95 reciprocal translocations of autosomes. Hum Genet. 1978;45:250–282. - PubMed
-
- Bier DM, Kaplan SL, Havel RJ. The Prader-Willi syndrome: Regulation of fat transport. Diabetes. 1977;26:874–881. - PubMed
-
- Bonuccelli CM, Stetten G, Levitt RC, Levin LS, Pyeritz RE. Prader-Willi syndrome associated with an interstitial deletion of chromosome 15. Johns Hopkins Med J. 1982;151(5):237–42. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical