

Hydroxyurea responsiveness in β-thalassemic patients is determined by the stress response adaptation of erythroid progenitors and their differentiation propensity
- PMID: 23100274
- PMCID: PMC3640112
- DOI: 10.3324/haematol.2012.074492
Hydroxyurea responsiveness in β-thalassemic patients is determined by the stress response adaptation of erythroid progenitors and their differentiation propensity
Abstract
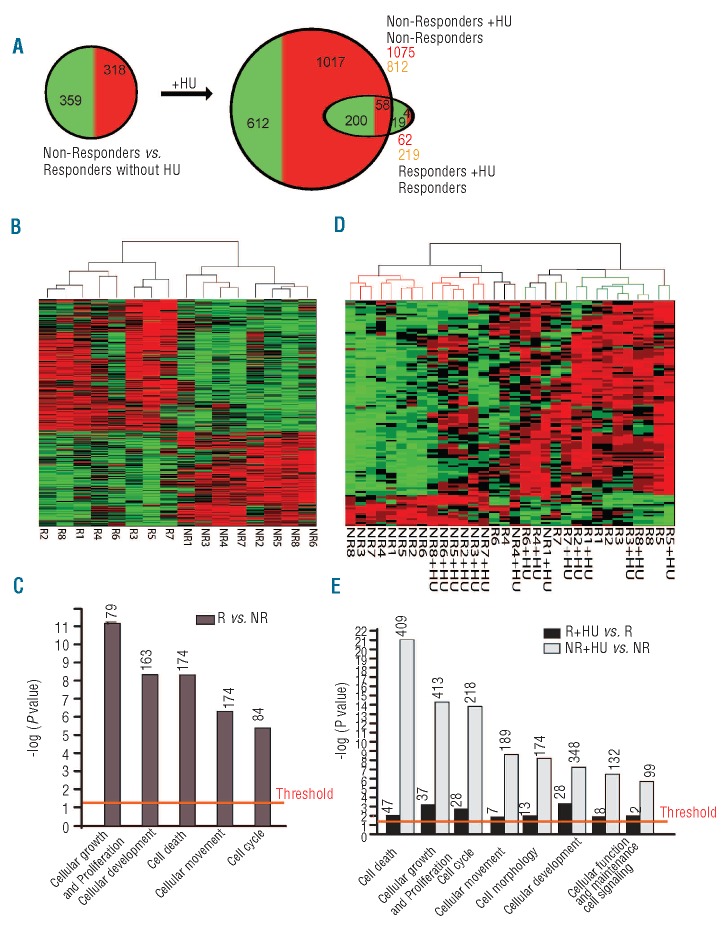
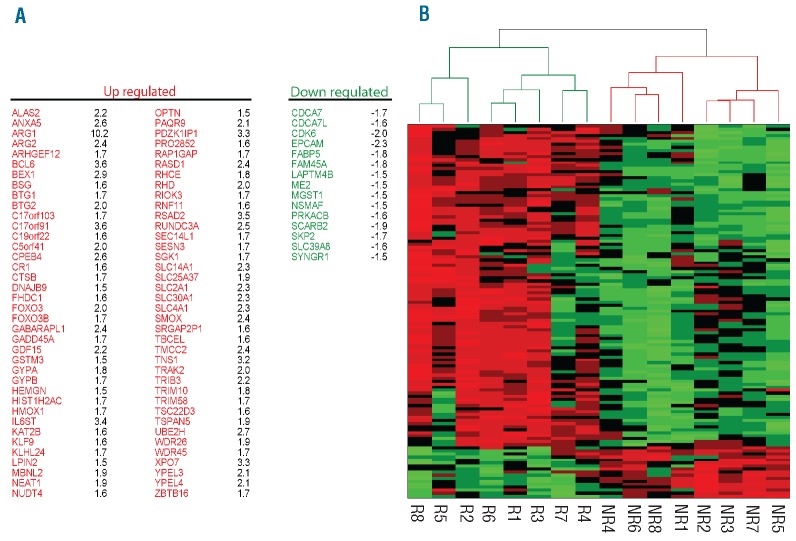
β-thalassemia is caused by mutations in the β-globin locus resulting in loss of, or reduced, hemoglobin A (adult hemoglobin, HbA, α2β2) production. Hydroxyurea treatment increases fetal γ-globin (fetal hemoglobin, HbF, α2γ2) expression in postnatal life substituting for the missing adult β-globin and is, therefore, an attractive therapeutic approach. Patients treated with hydroxyurea fall into three categories: i) 'responders' who increase hemoglobin to therapeutic levels; (ii) 'moderate-responders' who increase hemoglobin levels but still need transfusions at longer intervals; and (iii) 'non-responders' who do not reach adequate hemoglobin levels and remain transfusion-dependent. The mechanisms underlying these differential responses remain largely unclear. We generated RNA expression profiles from erythroblast progenitors of 8 responder and 8 non-responder β-thalassemia patients. These profiles revealed that hydroxyurea treatment induced differential expression of many genes in cells from non-responders while it had little impact on cells from responders. Part of the gene program up-regulated by hydroxyurea in non-responders was already highly expressed in responders before hydroxyurea treatment. Baseline HbF expression was low in non-responders, and hydroxyurea treatment induced significant cell death. We conclude that cells from responders have adapted well to constitutive stress conditions and display a propensity to proceed to the erythroid differentiation program.
Figures

Comment in
-
To respond or not to respond to hydroxyurea in thalassemia: a matter of stress adaptation?Haematologica. 2013 May;98(5):657-9. doi: 10.3324/haematol.2013.084392. Haematologica. 2013. PMID: 23633538 Free PMC article. No abstract available.
References
-
- Weatherall DJ, Clegg JB, Higgs DR, Wood WG. The hemoglobinopathies. In: Scriver CR, ed. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:4571–636
-
- Borgna-Pignatti C, Gamberini MR. Complications of thalassemia major and their treatment. Expert Rev Hematol. 2011; 4(3):353–66 - PubMed
-
- Borgna-Pignatti C, Rugolotto S, De Stefano P, Zhao H, Cappellini MD, Del Vecchio GC, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187–93 - PubMed
-
- Perrine SP, Miller BA, Greene MF, Cohen RA, Cook N, Shackleton C, et al. Butryic acid analogues augment gamma globin gene expression in neonatal erythroid progenitors. Bioche Biophys Res Commun. 1987;148(2):694–700 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
