

The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis
- PMID: 23100451
- PMCID: PMC3717962
- DOI: 10.18632/oncotarget.714
The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis
Abstract
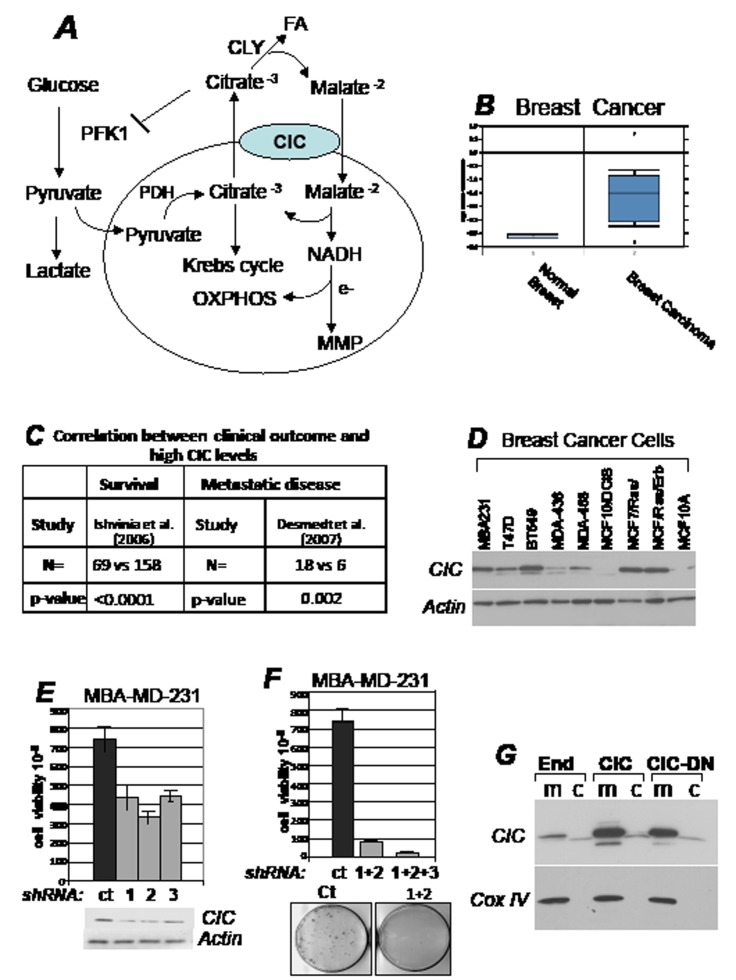
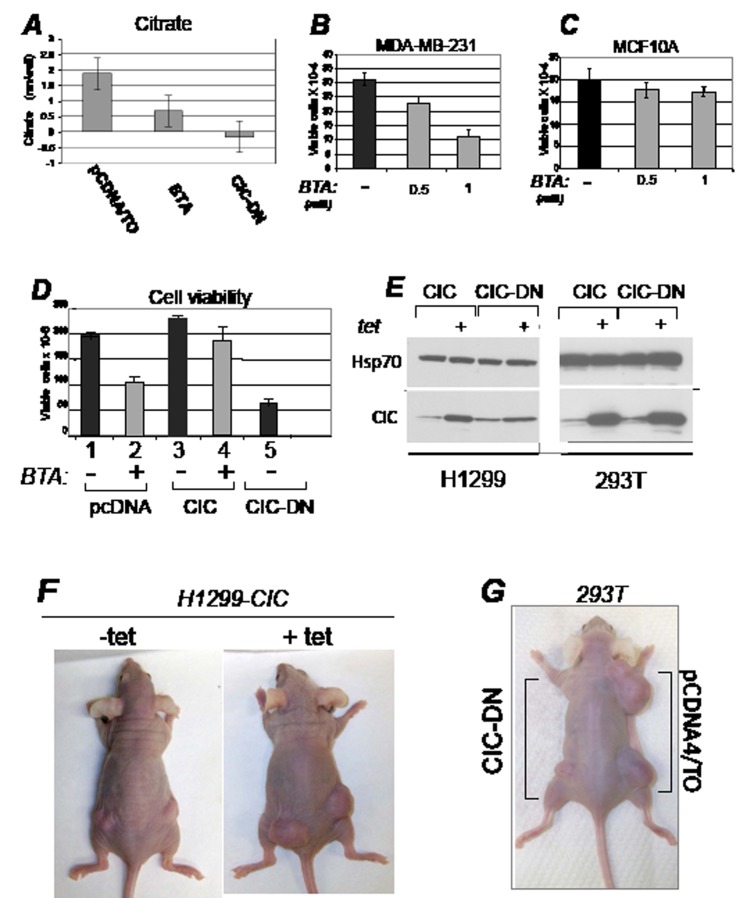
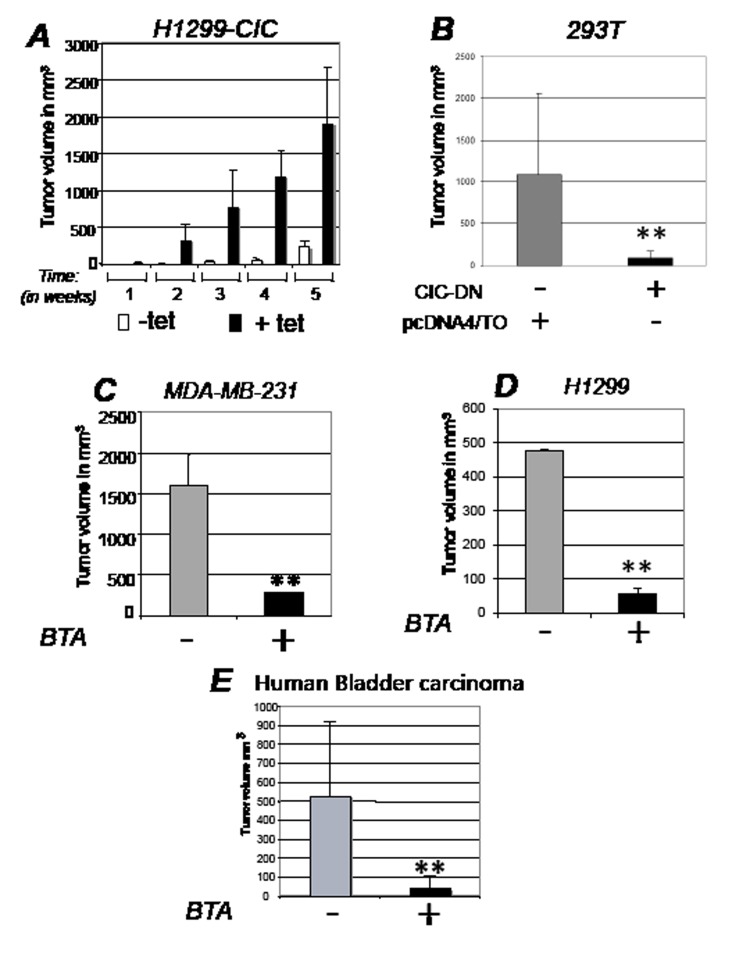
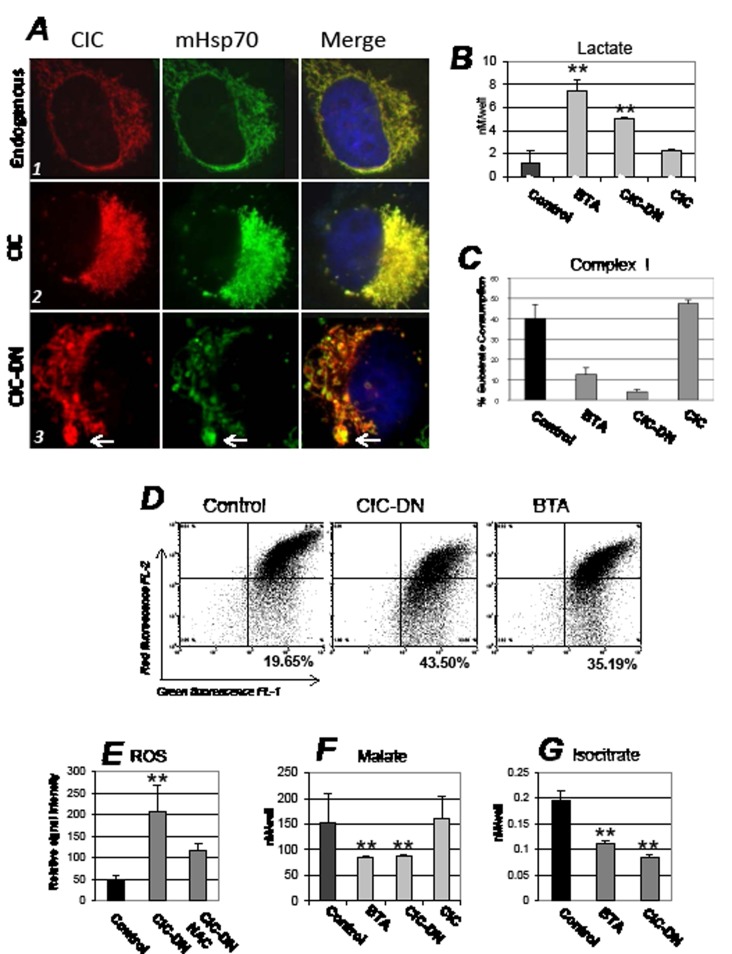
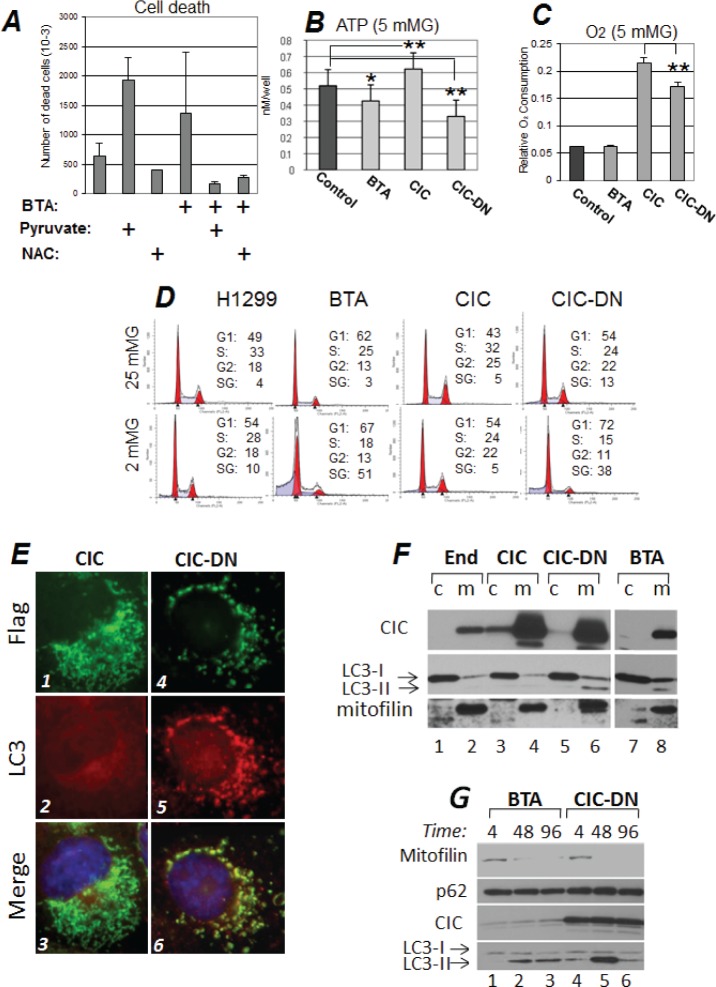
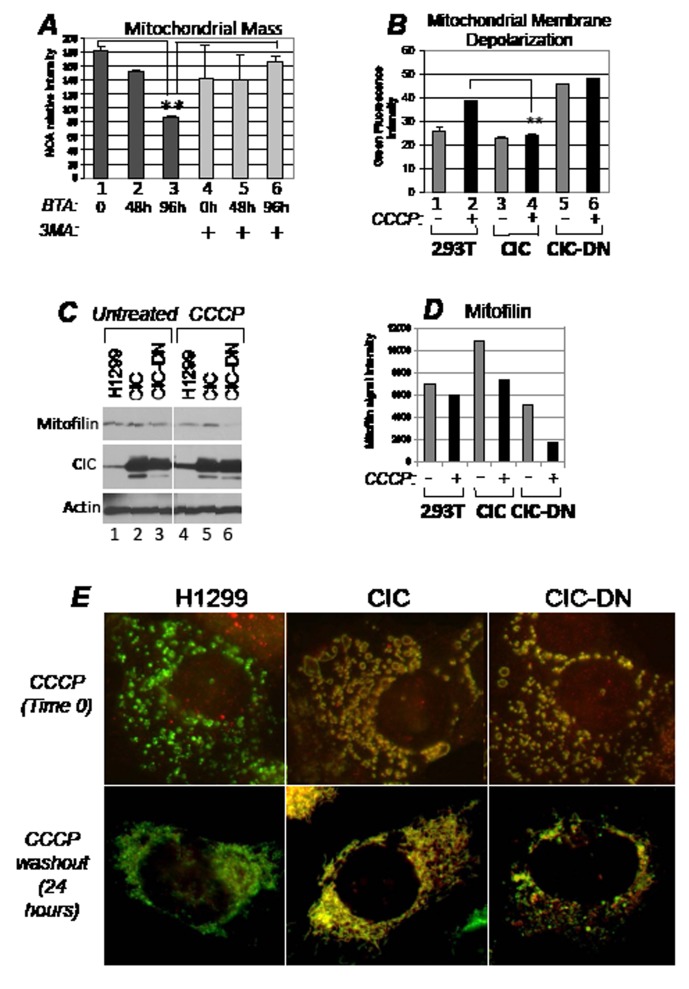
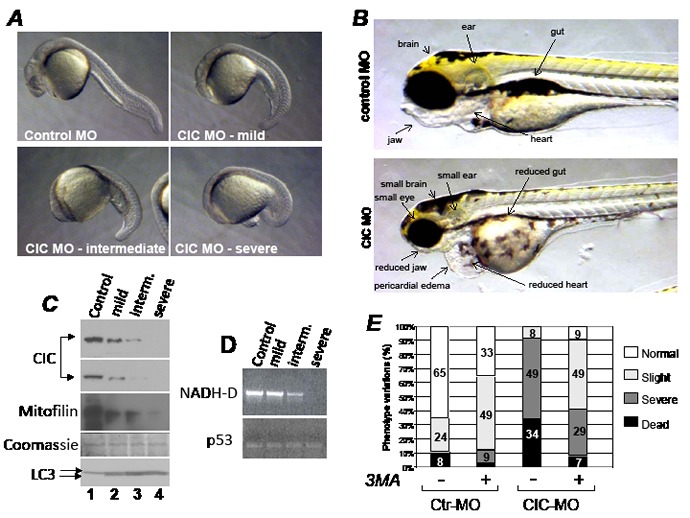
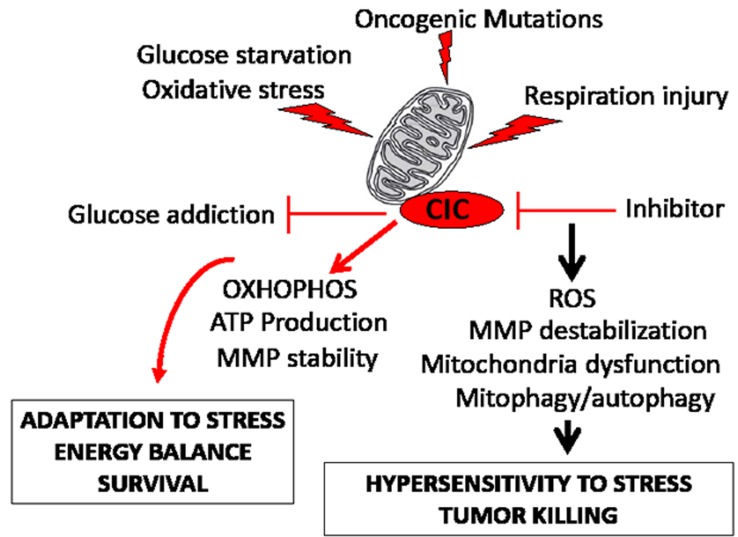
Dysregulation of the pathways that preserve mitochondrial integrity hallmarks many human diseases including diabetes, neurodegeration, aging and cancer. The mitochondrial citrate transporter gene, SLC25A1 or CIC, maps on chromosome 22q11.21, a region amplified in some tumors and deleted in developmental disorders known as velo-cardio-facial- and DiGeorge syndromes. We report here that in tumor cells CIC maintains mitochondrial integrity and bioenergetics, protects from mitochondrial damage and circumvents mitochondrial depletion via autophagy, hence promoting proliferation. CIC levels are increased in human cancers and its inhibition has anti-tumor activity, albeit with no toxicity on adult normal tissues. The knock-down of the CIC gene in zebrafish leads to mitochondria depletion and to proliferation defects that recapitulate features of human velo-cardio-facial syndrome, a phenotype rescued by blocking autophagy. Our findings reveal that CIC maintains mitochondrial homeostasis in metabolically active, high proliferating tissues and imply that this protein is a therapeutic target in cancer and likely, in other human diseases.
Figures
Comment in
-
Genetic induction of the Warburg effect inhibits tumor growth.Oncotarget. 2012 Nov;3(11):1266-7. doi: 10.18632/oncotarget.739. Oncotarget. 2012. PMID: 23175502 Free PMC article. No abstract available.
References
-
- Palmieri F, Pierri CL. Mitochondrial metabolite transport. Essays Biochem. 2010;47:37–52. - PubMed
-
- Nicholls DG. Mitochondrial ion circuits. Essays Biochem. 2010;47:25–35. - PubMed
-
- Palmieri F. Diseases caused by defects of mitochondrial carriers: a review. Biochim Biophys Acta. 2008;1777:564–578. - PubMed
-
- Stoffel M, Karayiorgou M, Espinosa R, 3rd, Beau MM. The human mitochondrial citrate transporter gene SLC20A3 maps to chromosome band 22q11 within a region implicated in DiGeorge syndrome, velo-cardio-facial syndrome and schizophrenia. Hum Genet. 1996;98:113–115. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
