

Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data
- PMID: 23103226
- PMCID: PMC3487130
- DOI: 10.1016/j.ajhg.2012.09.004
Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data
Abstract
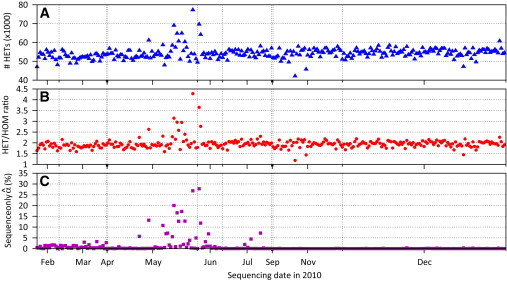
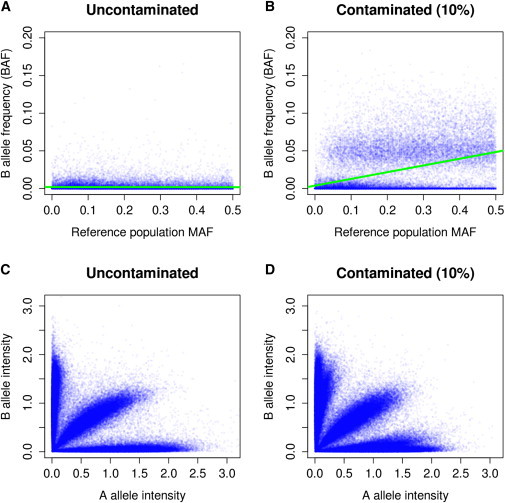
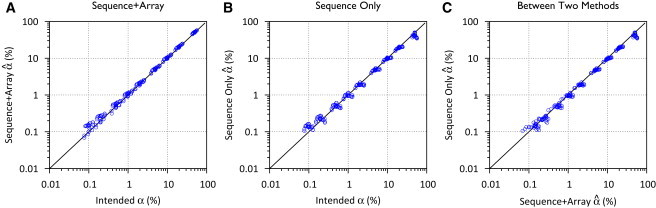
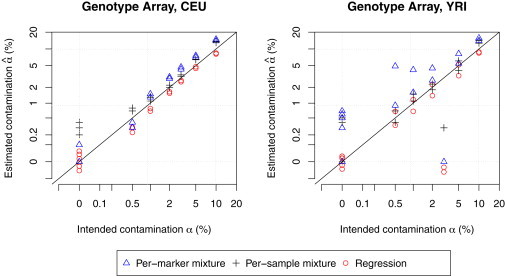
DNA sample contamination is a serious problem in DNA sequencing studies and may result in systematic genotype misclassification and false positive associations. Although methods exist to detect and filter out cross-species contamination, few methods to detect within-species sample contamination are available. In this paper, we describe methods to identify within-species DNA sample contamination based on (1) a combination of sequencing reads and array-based genotype data, (2) sequence reads alone, and (3) array-based genotype data alone. Analysis of sequencing reads allows contamination detection after sequence data is generated but prior to variant calling; analysis of array-based genotype data allows contamination detection prior to generation of costly sequence data. Through a combination of analysis of in silico and experimentally contaminated samples, we show that our methods can reliably detect and estimate levels of contamination as low as 1%. We evaluate the impact of DNA contamination on genotype accuracy and propose effective strategies to screen for and prevent DNA contamination in sequencing studies.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
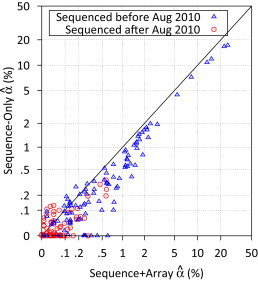
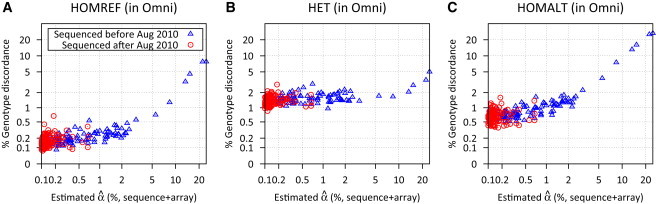
References
-
- Brent R.P. Dover Publications; New York: 2002. Algorithms for Minimization without Derivatives.
-
- Gordon D., Yang Y., Haynes C., Finch S.J., Mendell N.R., Brown A.M., Haroutunian V. Increasing power for tests of genetic association in the presence of phenotype and/or genotype error by use of double-sampling. Stat. Appl. Genet. Mol. Biol. 2004;3:e26. - PubMed
Publication types
MeSH terms
Grants and funding
- MH084698/MH/NIMH NIH HHS/United States
- R01 MH084698/MH/NIMH NIH HHS/United States
- T32 HG000040/HG/NHGRI NIH HHS/United States
- R56 HG000376/HG/NHGRI NIH HHS/United States
- R13 DK088398/DK/NIDDK NIH HHS/United States
- HHSN268200782096C/HG/NHGRI NIH HHS/United States
- HG006513/HG/NHGRI NIH HHS/United States
- HG005214/HG/NHGRI NIH HHS/United States
- P30 DK020572/DK/NIDDK NIH HHS/United States
- R01 HG007022/HG/NHGRI NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- HG000376/HG/NHGRI NIH HHS/United States
- DK088398/DK/NIDDK NIH HHS/United States
- U01 HG006513/HG/NHGRI NIH HHS/United States
- U01 HG005214/HG/NHGRI NIH HHS/United States
- HHSN268201100011I/HL/NHLBI NIH HHS/United States
- HHSN268201100011C/HL/NHLBI NIH HHS/United States
- R01 HG000376/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
