

In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome
- PMID: 23103230
- PMCID: PMC3487125
- DOI: 10.1016/j.ajhg.2012.10.002
In-frame mutations in exon 1 of SKI cause dominant Shprintzen-Goldberg syndrome
Abstract
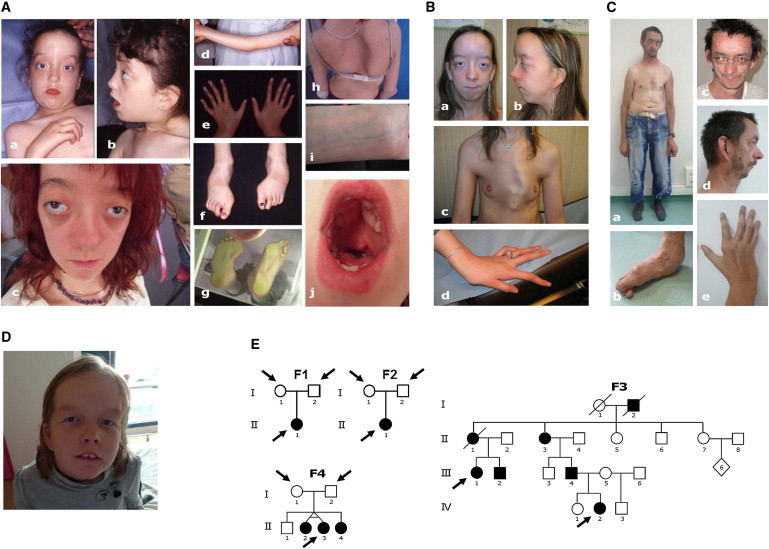
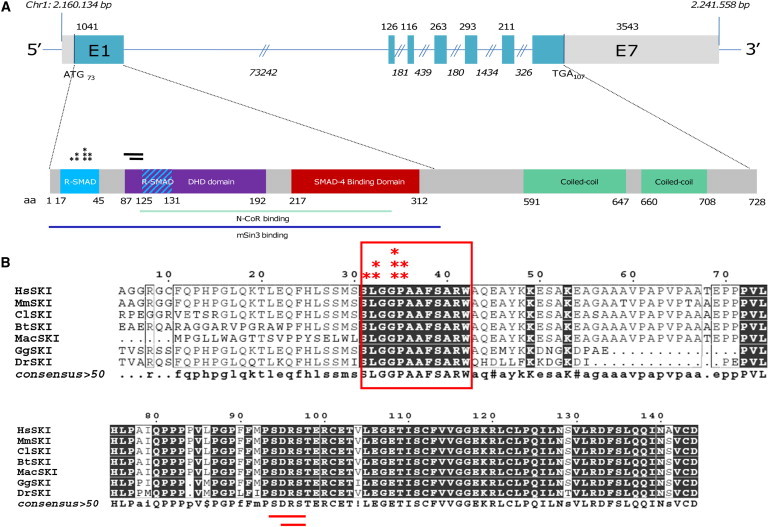
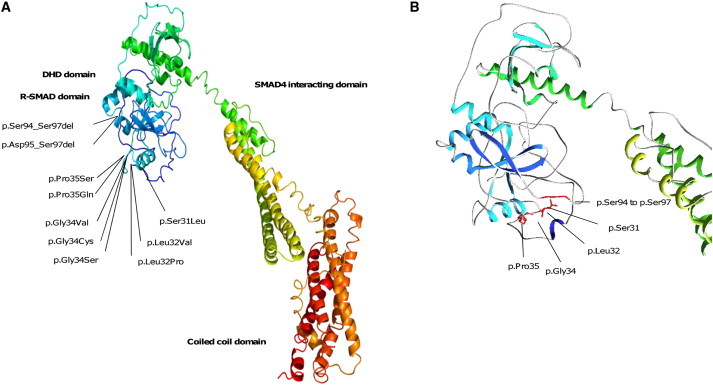
Shprintzen-Goldberg syndrome (SGS) is characterized by severe marfanoid habitus, intellectual disability, camptodactyly, typical facial dysmorphism, and craniosynostosis. Using family-based exome sequencing, we identified a dominantly inherited heterozygous in-frame deletion in exon 1 of SKI. Direct sequencing of SKI further identified one overlapping heterozygous in-frame deletion and ten heterozygous missense mutations affecting recurrent residues in 18 of the 19 individuals screened for SGS; these individuals included one family affected by somatic mosaicism. All mutations were located in a restricted area of exon 1, within the R-SMAD binding domain of SKI. No mutation was found in a cohort of 11 individuals with other marfanoid-craniosynostosis phenotypes. The interaction between SKI and Smad2/3 and Smad 4 regulates TGF-β signaling, and the pattern of anomalies in Ski-deficient mice corresponds to the clinical manifestations of SGS. These findings define SGS as a member of the family of diseases associated with the TGF-β-signaling pathway.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Robinson P.N., Neumann L.M., Demuth S., Enders H., Jung U., König R., Mitulla B., Müller D., Muschke P., Pfeiffer L. Shprintzen-Goldberg syndrome: fourteen new patients and a clinical analysis. Am. J. Med. Genet. A. 2005;135:251–262. - PubMed
-
- Shprintzen R.J., Goldberg R.B. A recurrent pattern syndrome of craniosynostosis associated with arachnodactyly and abdominal hernias. J. Craniofac. Genet. Dev. Biol. 1982;2:65–74. - PubMed
-
- Adès L.C., Morris L.L., Power R.G., Wilson M., Haan E.A., Bateman J.F., Milewicz D.M., Sillence D.O. Distinct skeletal abnormalities in four girls with Shprintzen-Goldberg syndrome. Am. J. Med. Genet. 1995;57:565–572. - PubMed
-
- Kosaki K., Takahashi D., Udaka T., Kosaki R., Matsumoto M., Ibe S., Isobe T., Tanaka Y., Takahashi T. Molecular pathology of Shprintzen-Goldberg syndrome. Am. J. Med. Genet. A. 2006;140:104–108. author reply 109–110. - PubMed
-
- Van Lierde K.M., Mortier G., Loeys B., Baudonck N., De Ley S., Marks L.A., Van Borsel J. Overall intelligibility, language, articulation, voice and resonance characteristics in a child with Shprintzen-Goldberg syndrome. Int. J. Pediatr. Otorhinolaryngol. 2007;71:721–728. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- HL-102924/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- HL-102926/HL/NHLBI NIH HHS/United States
- HL-102925/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-102923/HL/NHLBI NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-103010/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
