

Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency
- PMID: 23104095
- PMCID: PMC3514453
- DOI: 10.1038/ni.2457
Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency
Abstract
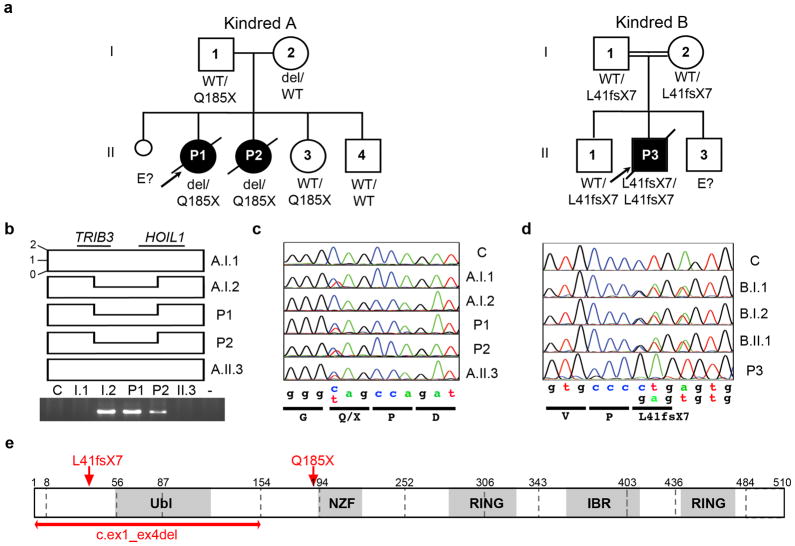
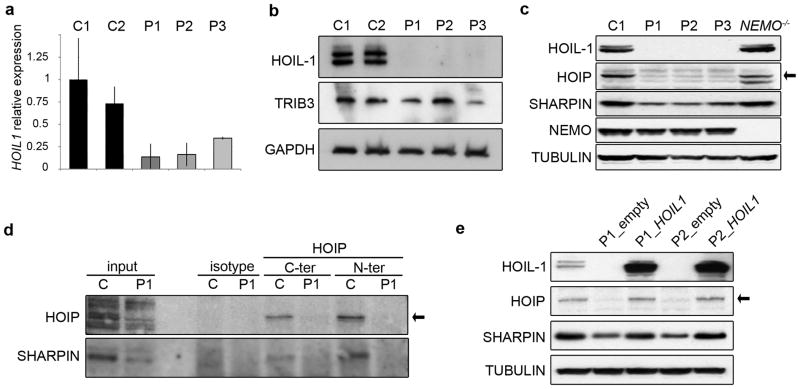
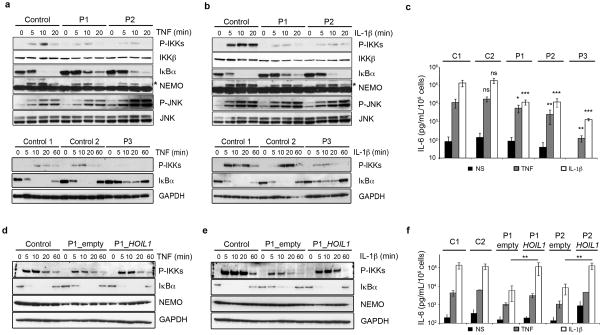
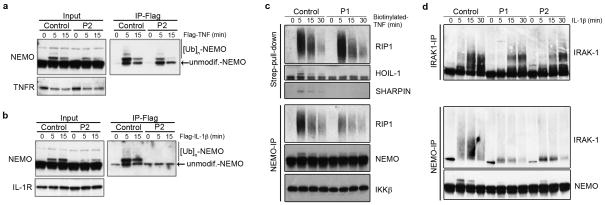
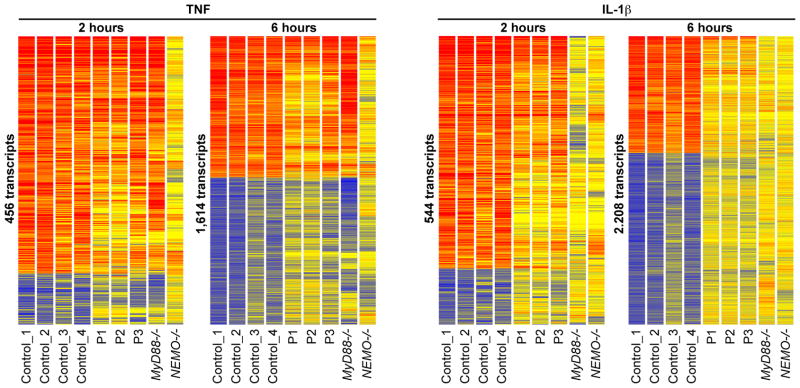
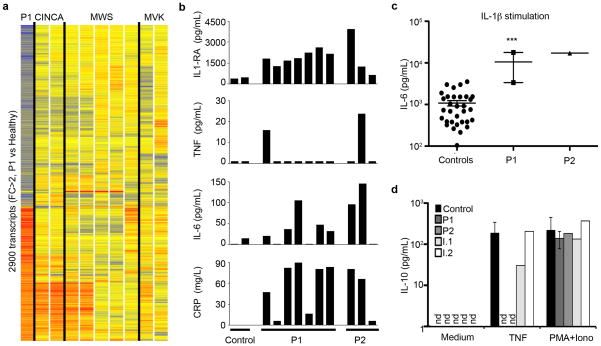
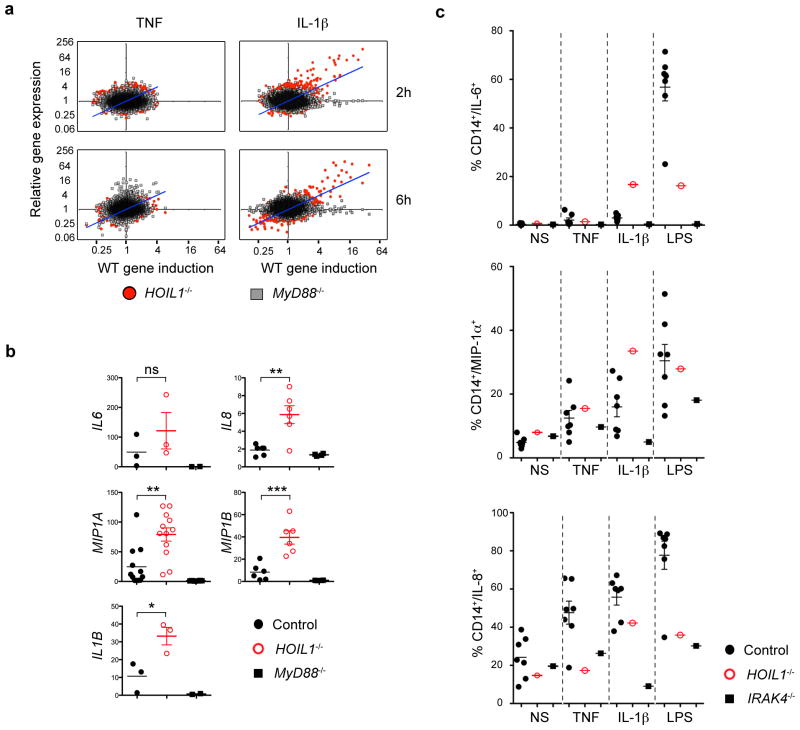
We report the clinical description and molecular dissection of a new fatal human inherited disorder characterized by chronic autoinflammation, invasive bacterial infections and muscular amylopectinosis. Patients from two kindreds carried biallelic loss-of-expression and loss-of-function mutations in HOIL1 (RBCK1), a component of the linear ubiquitination chain assembly complex (LUBAC). These mutations resulted in impairment of LUBAC stability. NF-κB activation in response to interleukin 1β (IL-1β) was compromised in the patients' fibroblasts. By contrast, the patients' mononuclear leukocytes, particularly monocytes, were hyper-responsive to IL-1β. The consequences of human HOIL-1 and LUBAC deficiencies for IL-1β responses thus differed between cell types, consistent with the unique association of autoinflammation and immunodeficiency in these patients. These data suggest that LUBAC regulates NF-κB-dependent IL-1β responses differently in different cell types.
Conflict of interest statement
Figures
Comment in
-
HOIL and water: the two faces of HOIL-1 deficiency.Nat Immunol. 2012 Dec;13(12):1133-5. doi: 10.1038/ni.2471. Nat Immunol. 2012. PMID: 23160206 No abstract available.
References
-
- McDermott MF, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–144. - PubMed
-
- Puel A, Picard C, Ku CL, Smahi A, Casanova JL. Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol. 2004;16:34–41. - PubMed
-
- Doffinger R, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–285. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
