

Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics
- PMID: 23107649
- PMCID: PMC3501970
- DOI: 10.1093/brain/aws231
Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics
Abstract
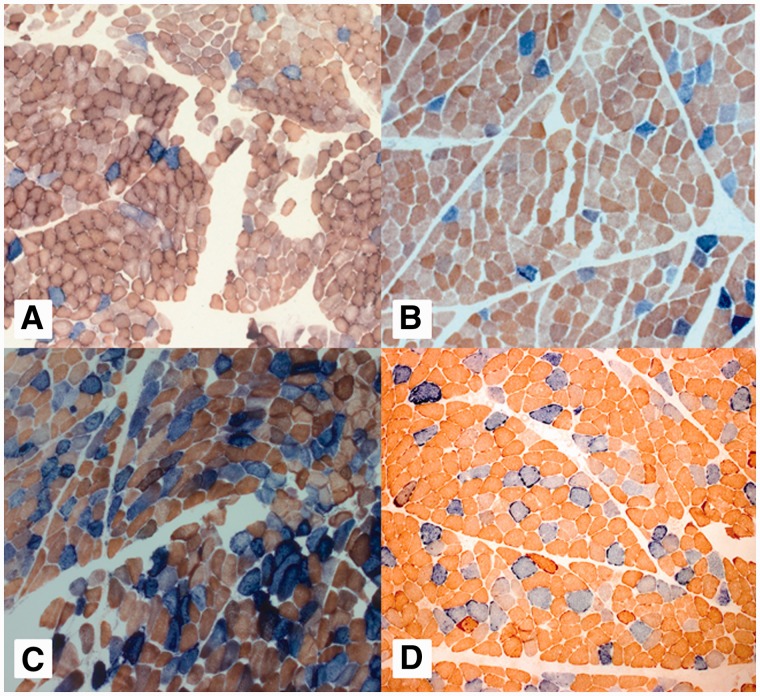
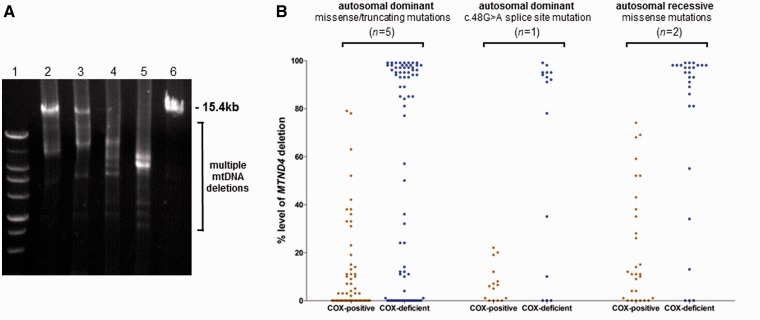
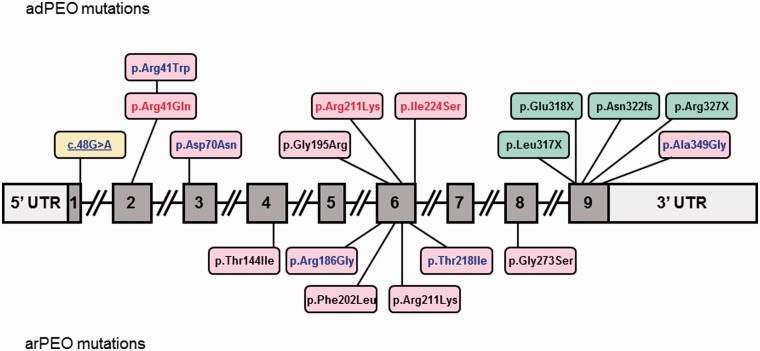
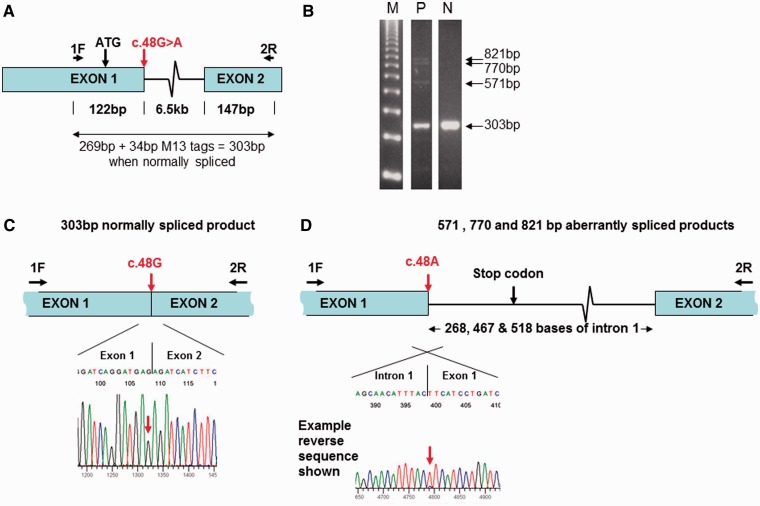
Mutations in the nuclear-encoded mitochondrial maintenance gene RRM2B are an important cause of familial mitochondrial disease in both adults and children and represent the third most common cause of multiple mitochondrial DNA deletions in adults, following POLG [polymerase (DNA directed), gamma] and PEO1 (now called C10ORF2, encoding the Twinkle helicase) mutations. However, the clinico-pathological and molecular features of adults with RRM2B-related disease have not been clearly defined. In this multicentre study of 26 adult patients from 22 independent families, including five additional cases published in the literature, we show that extra-ocular neurological complications are common in adults with genetically confirmed RRM2B mutations. We also demonstrate a clear correlation between the clinical phenotype and the underlying genetic defect. Myopathy was a prominent manifestation, followed by bulbar dysfunction and fatigue. Sensorineural hearing loss and gastrointestinal disturbance were also important findings. Severe multisystem neurological disease was associated with recessively inherited compound heterozygous mutations with a mean age of disease onset at 7 years. Dominantly inherited heterozygous mutations were associated with a milder predominantly myopathic phenotype with a later mean age of disease onset at 46 years. Skeletal muscle biopsies revealed subsarcolemmal accumulation of mitochondria and/or cytochrome c oxidase-deficient fibres. Multiple mitochondrial DNA deletions were universally present in patients who underwent a muscle biopsy. We identified 18 different heterozygous RRM2B mutations within our cohort of patients, including five novel mutations that have not previously been reported. Despite marked clinical overlap between the mitochondrial maintenance genes, key clinical features such as bulbar dysfunction, hearing loss and gastrointestinal disturbance should help prioritize genetic testing towards RRM2B analysis, and sequencing of the gene may preclude performance of a muscle biopsy.
Figures
References
-
- Acham-Roschitz B, Plecko B, Lindbichler F, Bittner R, Mache CJ, Sperl W, et al. A novel mutation of the RRM2B gene in an infant with early fatal encephalomyopathy, central hypomyelination, and tubulopathy. Mol Genet Metab. 2009;98:300–4. - PubMed
-
- Blakely E, He L, Gardner JL, Hudson G, Walter J, Hughes I, et al. Novel mutations in the TK2 gene associated with fatal mitochondrial DNA depletion myopathy. Neuromuscul Disord. 2008;18:557–60. - PubMed
-
- Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–80. - PubMed
