

Defective autophagy and mTORC1 signaling in myotubularin null mice
- PMID: 23109424
- PMCID: PMC3536306
- DOI: 10.1128/MCB.01075-12
Defective autophagy and mTORC1 signaling in myotubularin null mice
Abstract
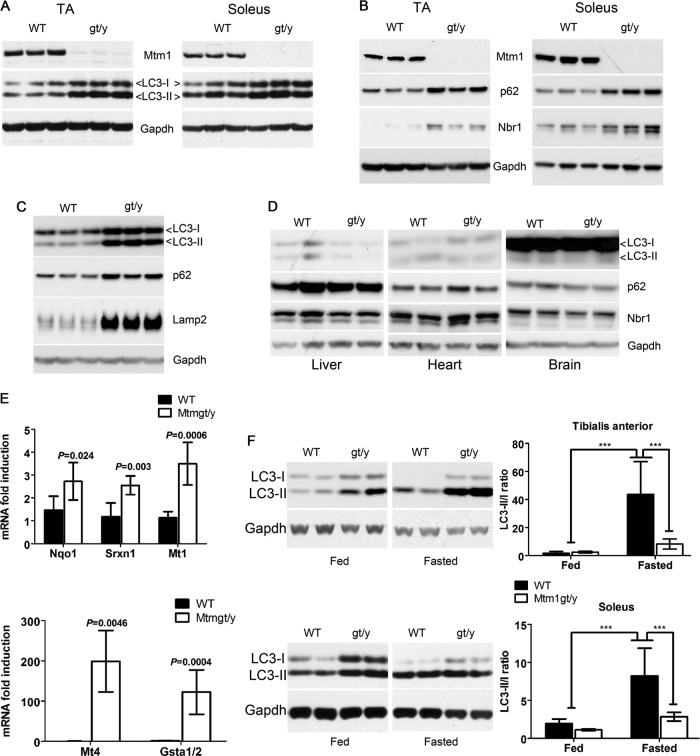
Autophagy is a vesicular trafficking pathway that regulates the degradation of aggregated proteins and damaged organelles. Initiation of autophagy requires several multiprotein signaling complexes, such as the ULK1 kinase complex and the Vps34 lipid kinase complex, which generates phosphatidylinositol 3-phosphate [PtdIns(3)P] on the forming autophagosomal membrane. Alterations in autophagy have been reported for various diseases, including myopathies. Here we show that skeletal muscle autophagy is compromised in mice deficient in the X-linked myotubular myopathy (XLMTM)-associated PtdIns(3)P phosphatase myotubularin (MTM1). Mtm1-deficient muscle displays several cellular abnormalities, including a profound increase in ubiquitin aggregates and abnormal mitochondria. Further, we show that Mtm1 deficiency is accompanied by activation of mTORC1 signaling, which persists even following starvation. In vivo pharmacological inhibition of mTOR is sufficient to normalize aberrant autophagy and improve muscle phenotypes in Mtm1 null mice. These results suggest that aberrant mTORC1 signaling and impaired autophagy are consequences of the loss of Mtm1 and may play a primary role in disease pathogenesis.
Figures







References
-
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Mishima K, Saito I, Okano H, Mizushima N. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885– 889 - PubMed
-
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880– 884 - PubMed
-
- Komatsu M, Waguri S, Koike M, Sou Y, Ueno T, Hara T, Mizushima N, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131: 1149– 1163 - PubMed
-
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. 2007. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13: 619– 624 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous