

Analysis of a wild mouse promoter variant reveals a novel role for FcγRIIb in the control of the germinal center and autoimmunity
- PMID: 23109709
- PMCID: PMC3501356
- DOI: 10.1084/jem.20121752
Analysis of a wild mouse promoter variant reveals a novel role for FcγRIIb in the control of the germinal center and autoimmunity
Abstract
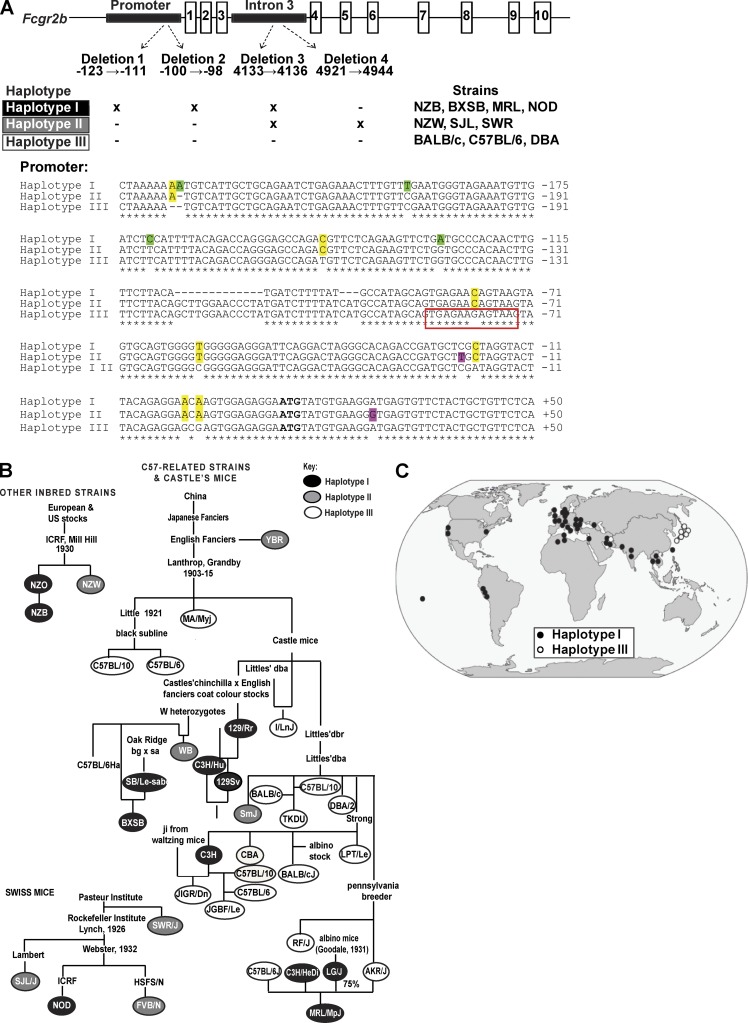
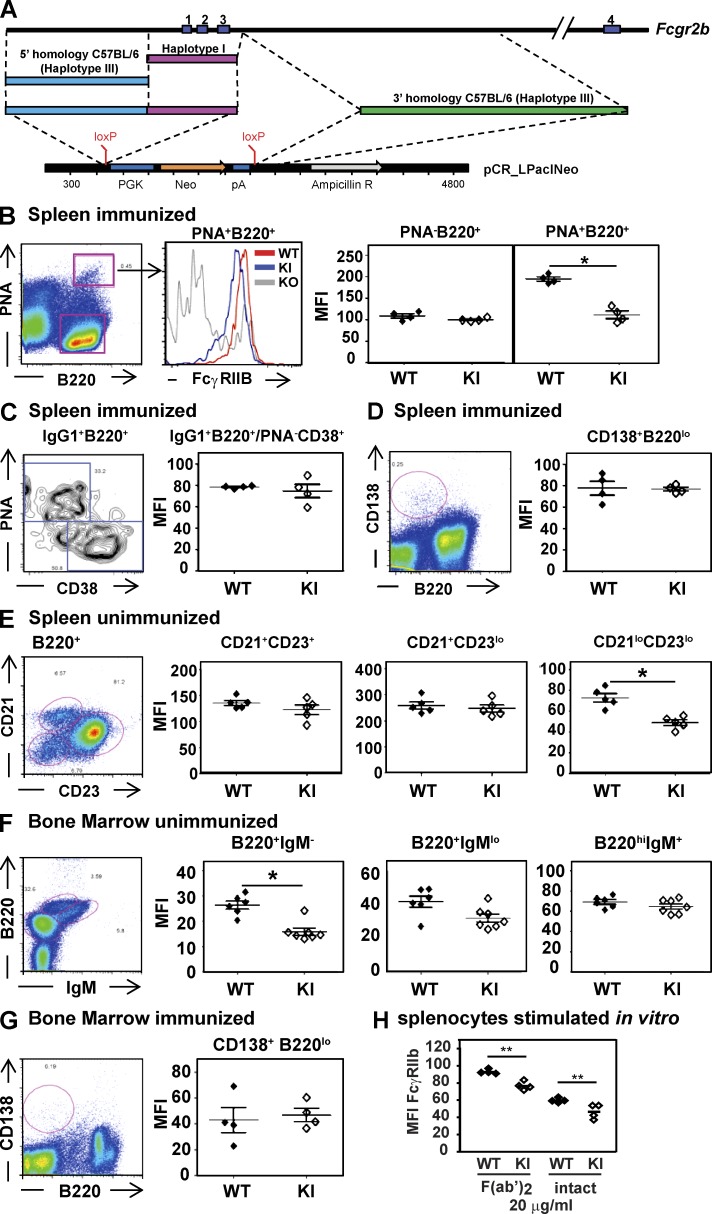
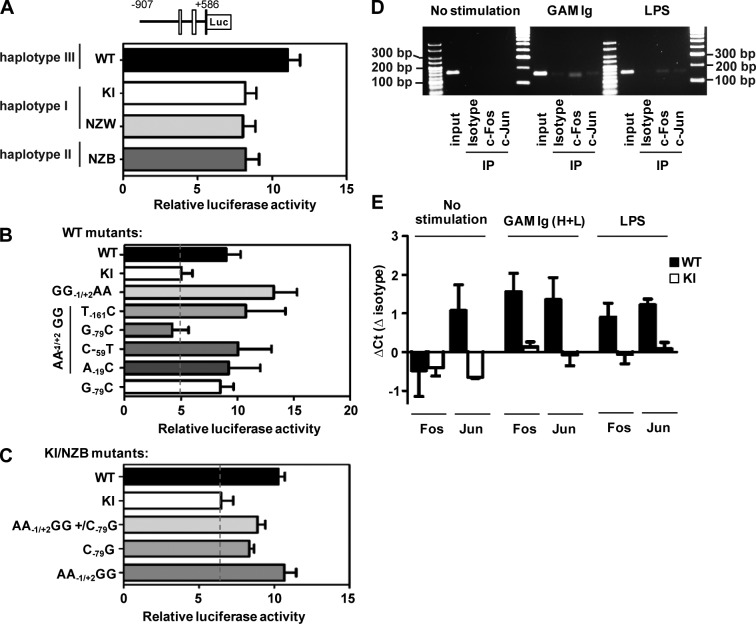
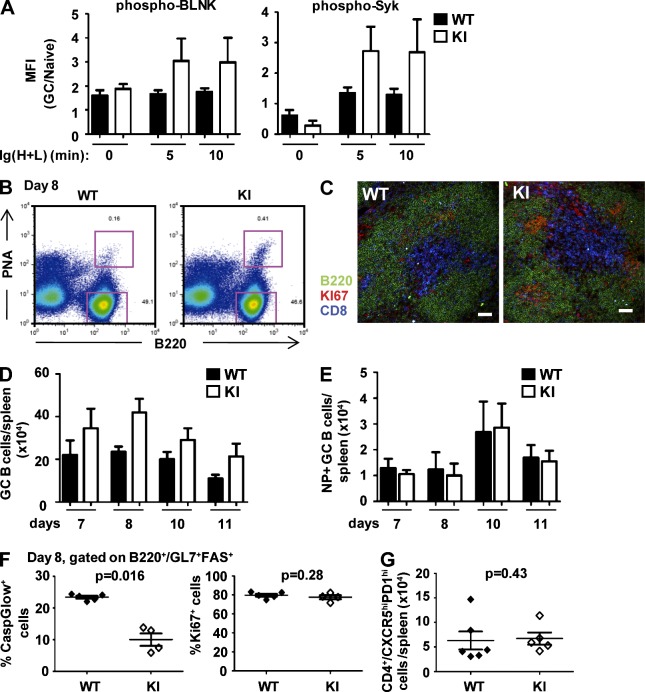
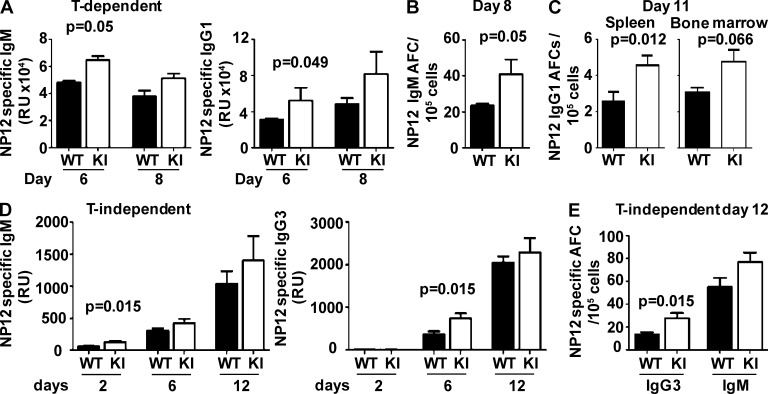
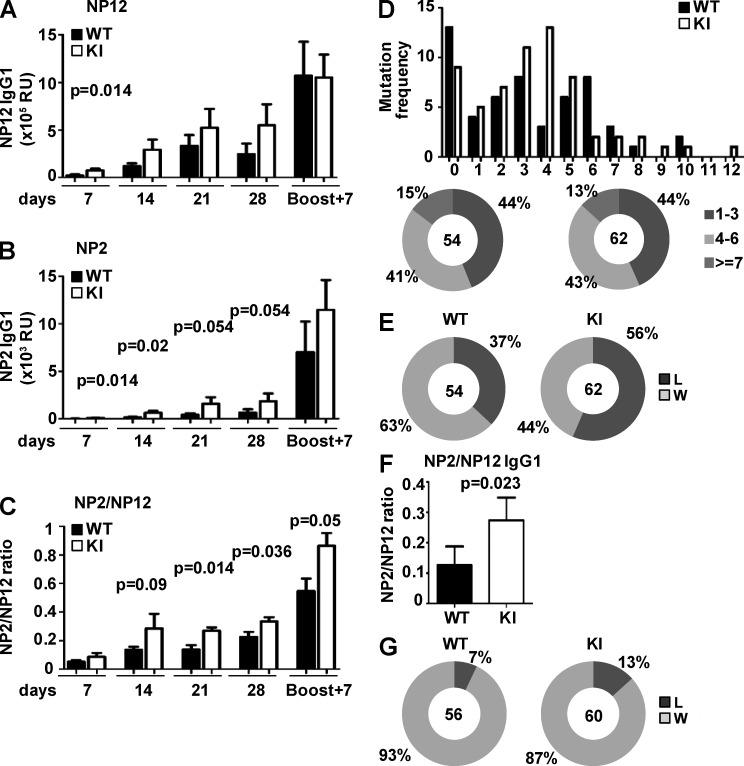
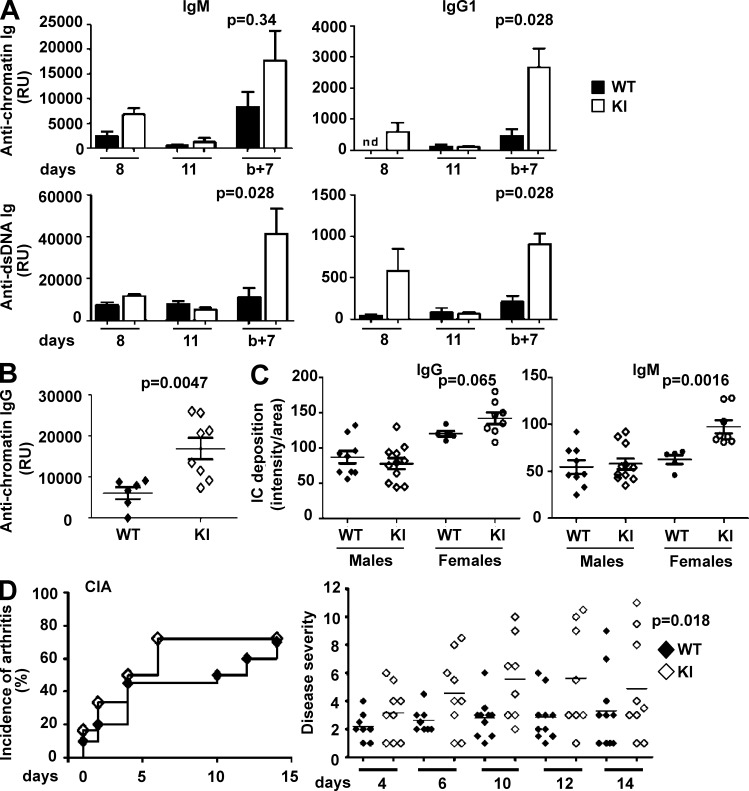
Genetic variants of the inhibitory Fc receptor FcγRIIb have been associated with systemic lupus erythematosus in humans and mice. The mechanism by which Fcgr2b variants contribute to the development of autoimmunity is unknown and was investigated by knocking in the most commonly conserved wild mouse Fcgr2b promoter haplotype, also associated with autoimmune-prone mouse strains, into the C57BL/6 background. We found that in the absence of an AP-1-binding site in its promoter, FcγRIIb failed to be up-regulated on activated and germinal center (GC) B cells. This resulted in enhanced GC responses, increased affinity maturation, and autoantibody production. Accordingly, in the absence of FcγRIIb activation-induced up-regulation, mice developed more severe collagen-induced arthritis and spontaneous glomerular immune complex deposition. Our data highlight how natural variation in Fcgr2b drives the development of autoimmune disease. They also show how the study of such variants using a knockin approach can provide insight into immune mechanisms not possible using conventional genetic manipulation, in this case demonstrating an unexpected critical role for the activation-induced up-regulation of FcγRIIb in controlling affinity maturation, autoantibody production, and autoimmunity.
Figures
Comment in
-
B cell responses: Born to be (a bit) wild.Nat Rev Immunol. 2012 Dec;12(12):808-9. doi: 10.1038/nri3347. Epub 2012 Nov 16. Nat Rev Immunol. 2012. PMID: 23154225 No abstract available.
References
-
- Blank M.C., Stefanescu R.N., Masuda E., Marti F., King P.D., Redecha P.B., Wurzburger R.J., Peterson M.G., Tanaka S., Pricop L. 2005. Decreased transcription of the human FCGR2B gene mediated by the -343 G/C promoter polymorphism and association with systemic lupus erythematosus. Hum. Genet. 117:220–227 10.1007/s00439-005-1302-3 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
