

Paneth cell-mediated multiorgan dysfunction after acute kidney injury
- PMID: 23109723
- PMCID: PMC3504173
- DOI: 10.4049/jimmunol.1200581
Paneth cell-mediated multiorgan dysfunction after acute kidney injury
Abstract
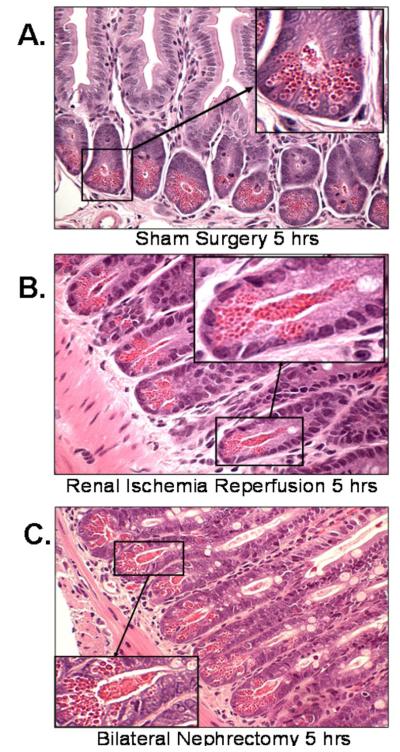
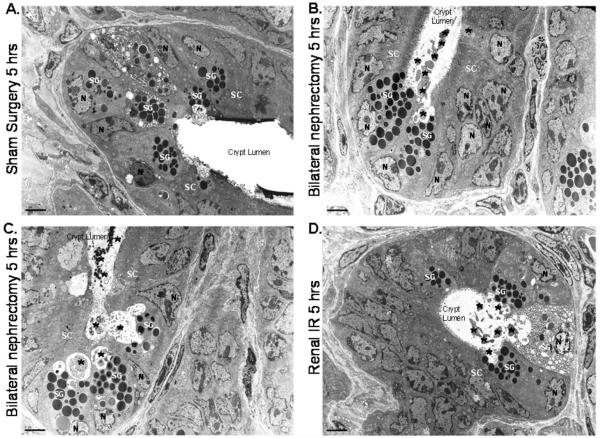
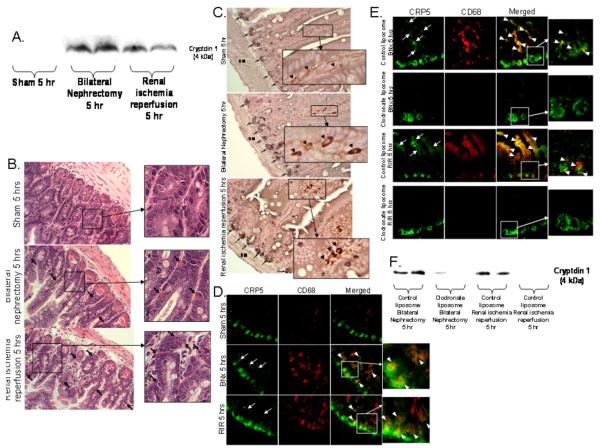
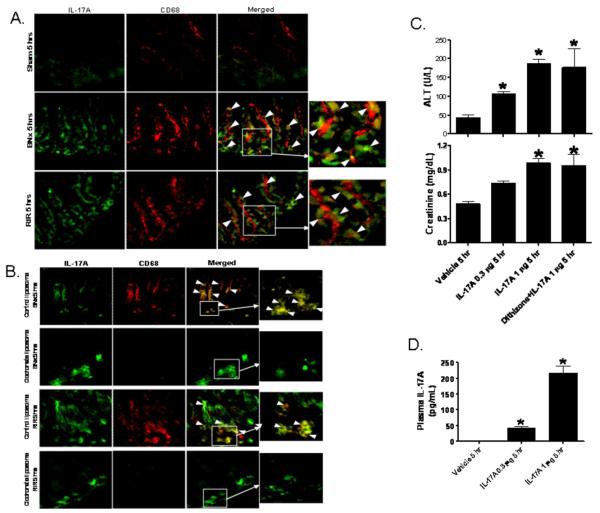
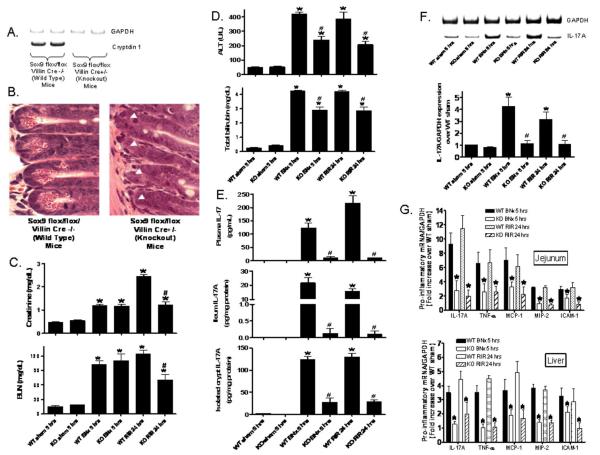
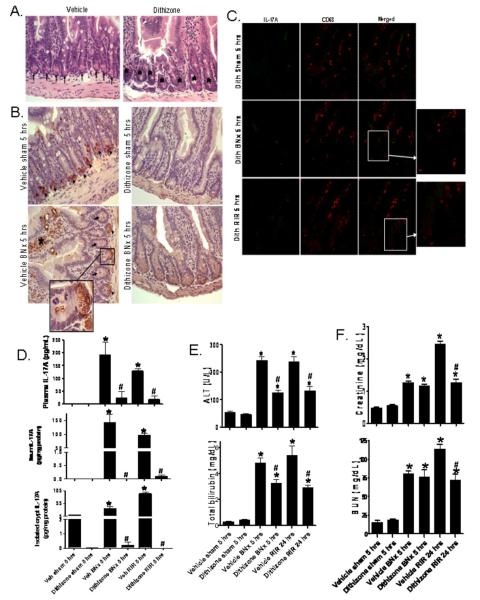
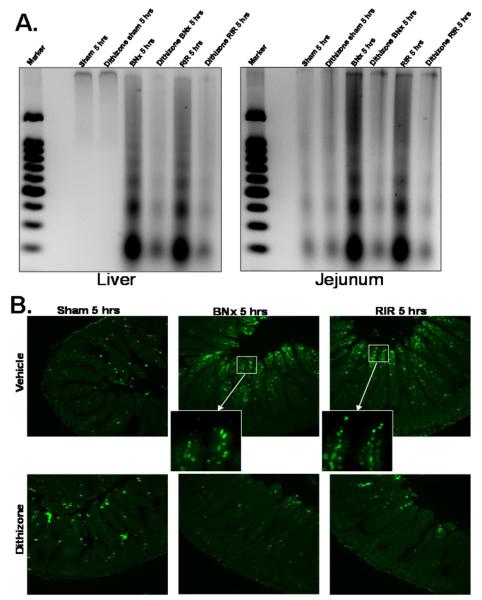
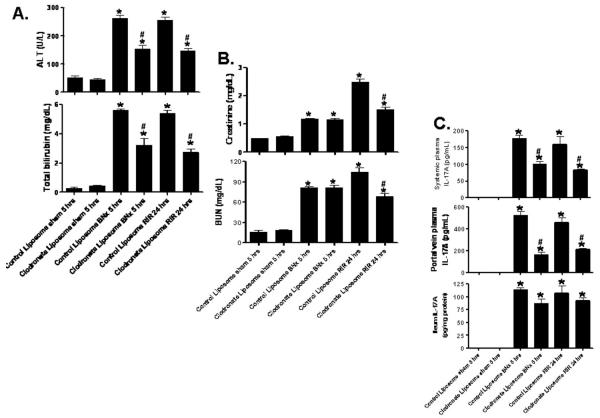
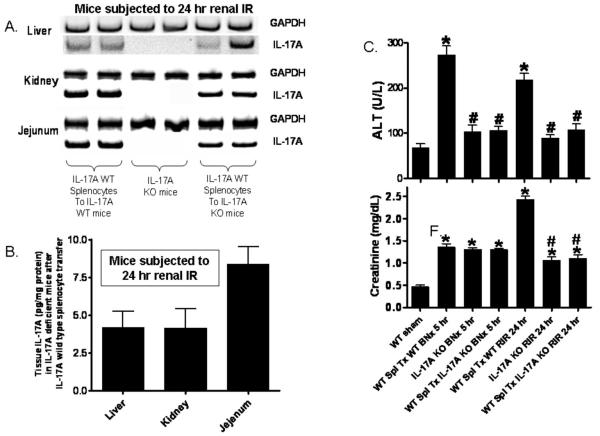
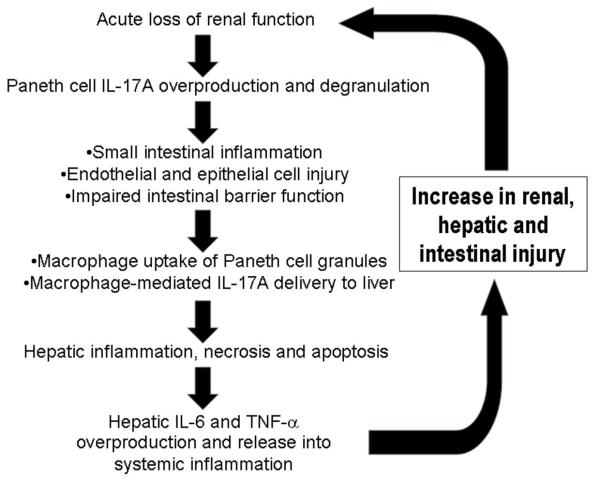
Acute kidney injury (AKI) is frequently complicated by extrarenal multiorgan injury, including intestinal and hepatic dysfunction. In this study, we hypothesized that a discrete intestinal source of proinflammatory mediators drives multiorgan injury in response to AKI. After induction of AKI in mice by renal ischemia-reperfusion or bilateral nephrectomy, small intestinal Paneth cells increased the synthesis and release of IL-17A in conjunction with severe intestinal apoptosis and inflammation. We also detected significantly increased IL-17A in portal and systemic circulation after AKI. Intestinal macrophages appear to transport released Paneth cell granule constituents induced by AKI, away from the base of the crypts into the liver. Genetic or pharmacologic depletion of Paneth cells decreased small intestinal IL-17A secretion and plasma IL-17A levels significantly and attenuated intestinal, hepatic, and renal injury after AKI. Similarly, portal delivery of IL-17A in macrophage-depleted mice decreased markedly. In addition, intestinal, hepatic, and renal injury following AKI was attenuated without affecting intestinal IL-17A generation. In conclusion, AKI induces IL-17A synthesis and secretion by Paneth cells to initiate intestinal and hepatic injury by hepatic and systemic delivery of IL-17A by macrophages. Modulation of Paneth cell dysregulation may have therapeutic implications by reducing systemic complications arising from AKI.
Figures
References
-
- Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephrol. 2005;16:3365. - PubMed
-
- Jones DR, Lee HT. Perioperative renal protection. Best. Pract. Res. Clin. Anaesthesiol. 2008;22:193. - PubMed
-
- Elapavaluru S, Kellum JA. Why do patients die of acute kidney injury? Acta Clin. Belg. 2007;(Suppl):326. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
