

The heterochromatic barrier to DNA double strand break repair: how to get the entry visa
- PMID: 23109886
- PMCID: PMC3472778
- DOI: 10.3390/ijms130911844
The heterochromatic barrier to DNA double strand break repair: how to get the entry visa
Abstract
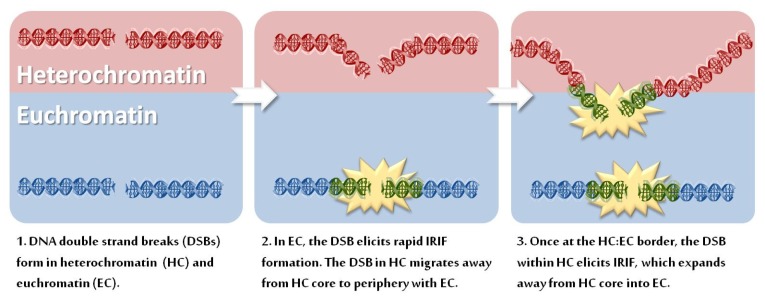
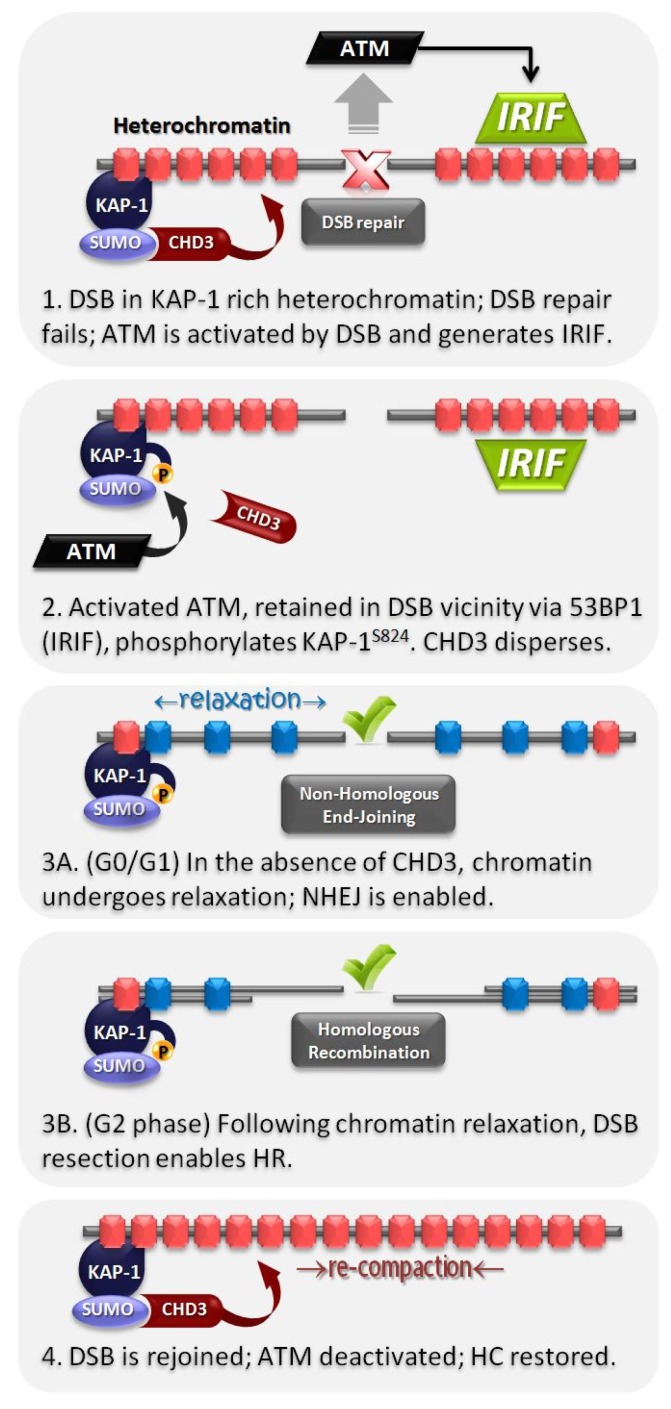
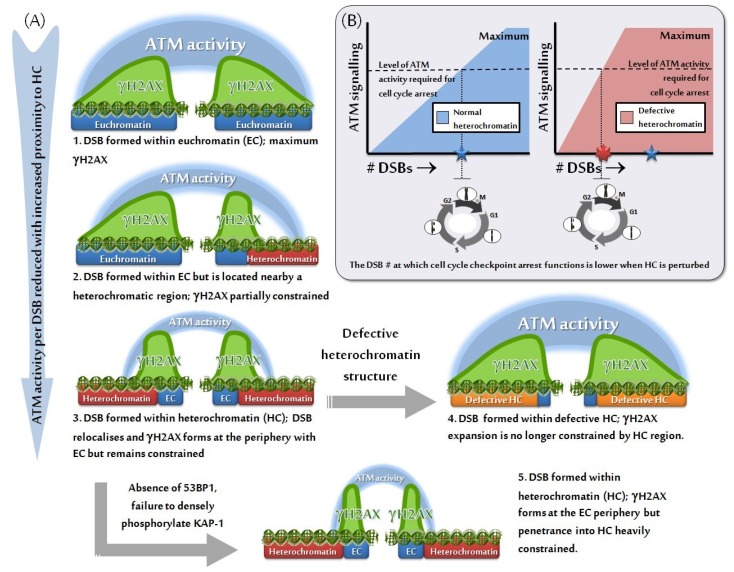
Over recent decades, a deep understanding of pathways that repair DNA double strand breaks (DSB) has been gained from biochemical, structural, biophysical and cellular studies. DNA non-homologous end-joining (NHEJ) and homologous recombination (HR) represent the two major DSB repair pathways, and both processes are now well understood. Recent work has demonstrated that the chromatin environment at a DSB significantly impacts upon DSB repair and that, moreover, dramatic modifications arise in the chromatin surrounding a DSB. Chromatin is broadly divided into open, transcriptionally active, euchromatin (EC) and highly compacted, transcriptionally inert, heterochromatin (HC), although these represent extremes of a spectrum. The HC superstructure restricts both DSB repair and damage response signaling. Moreover, DSBs within HC (HC-DSBs) are rapidly relocalized to the EC-HC interface. The damage response protein kinase, ataxia telangiectasia mutated (ATM), is required for HC-DSB repair but is dispensable for the relocalization of HC-DSBs. It has been proposed that ATM signaling enhances HC relaxation in the DSB vicinity and that this is a prerequisite for HC-DSB repair. Hence, ATM is essential for repair of HC-DSBs. Here, we discuss how HC impacts upon the response to DSBs and how ATM overcomes the barrier that HC poses to repair.
Keywords: DNA non-homologous end-joining; ataxia telangiectasia mutated; chromatin; damage response signaling; heterochromatin; homologous recombination.
Figures
References
-
- Ayoub N., Jeyasekharan A.D., Bernal J.A., Venkitaraman A.R. HP1-β mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–686. - PubMed
-
- Ayoub N., Jeyasekharan A.D., Venkitaraman A.R. Mobilization and recruitment of HP1: A bimodal response to DNA breakage. Cell Cycle. 2009;8:2945–2950. - PubMed
-
- Kasparek T.R., Humphrey T.C. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin. Cell Dev. Biol. 2011;22:886–897. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
