

Denoising PCR-amplified metagenome data
- PMID: 23113967
- PMCID: PMC3563472
- DOI: 10.1186/1471-2105-13-283
Denoising PCR-amplified metagenome data
Abstract
Background: PCR amplification and high-throughput sequencing theoretically enable the characterization of the finest-scale diversity in natural microbial and viral populations, but each of these methods introduces random errors that are difficult to distinguish from genuine biological diversity. Several approaches have been proposed to denoise these data but lack either speed or accuracy.
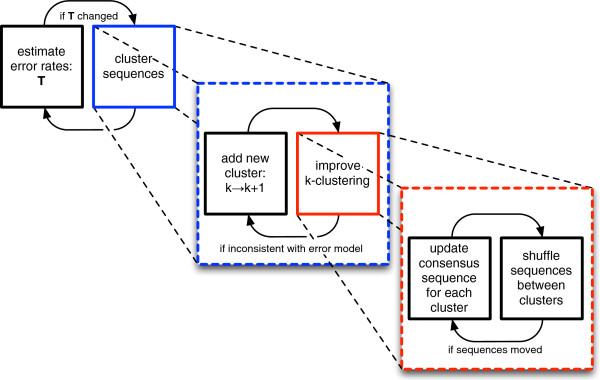
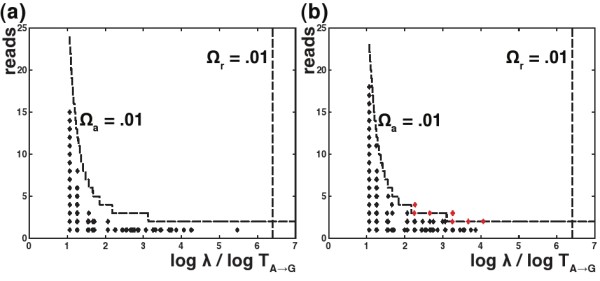
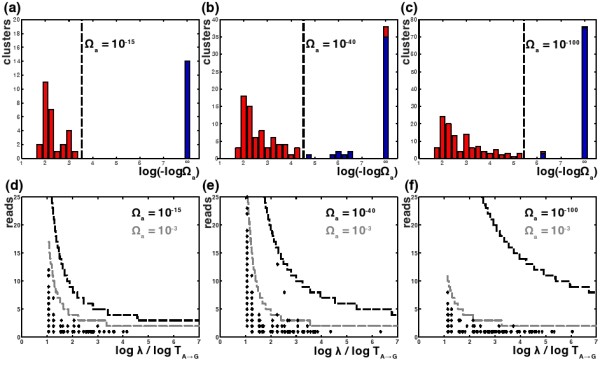
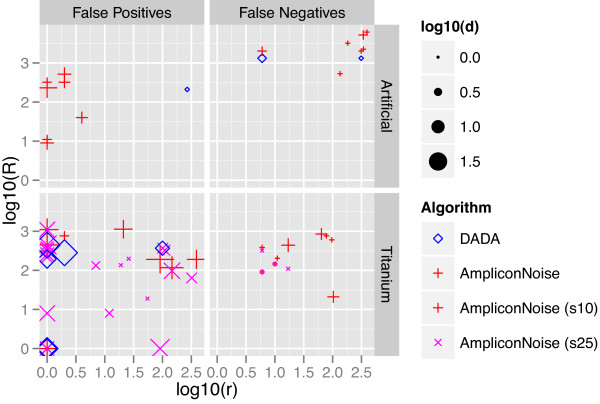
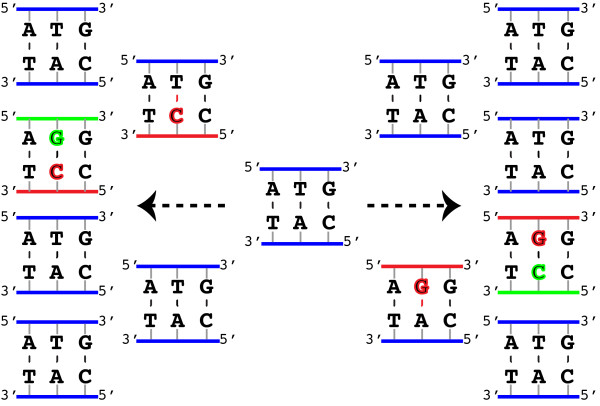
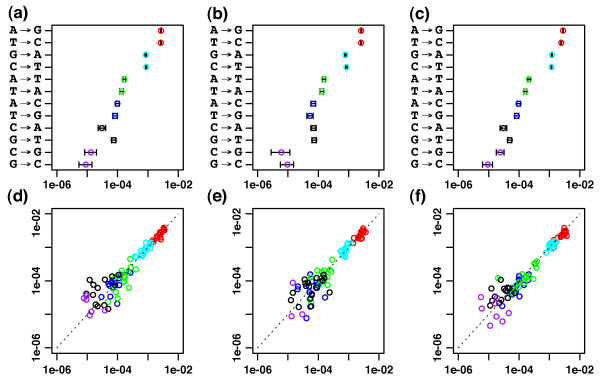
Results: We introduce a new denoising algorithm that we call DADA (Divisive Amplicon Denoising Algorithm). Without training data, DADA infers both the sample genotypes and error parameters that produced a metagenome data set. We demonstrate performance on control data sequenced on Roche's 454 platform, and compare the results to the most accurate denoising software currently available, AmpliconNoise.
Conclusions: DADA is more accurate and over an order of magnitude faster than AmpliconNoise. It eliminates the need for training data to establish error parameters, fully utilizes sequence-abundance information, and enables inclusion of context-dependent PCR error rates. It should be readily extensible to other sequencing platforms such as Illumina.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
