

DGAT1 mutation is linked to a congenital diarrheal disorder
- PMID: 23114594
- PMCID: PMC3533555
- DOI: 10.1172/JCI64873
DGAT1 mutation is linked to a congenital diarrheal disorder
Abstract
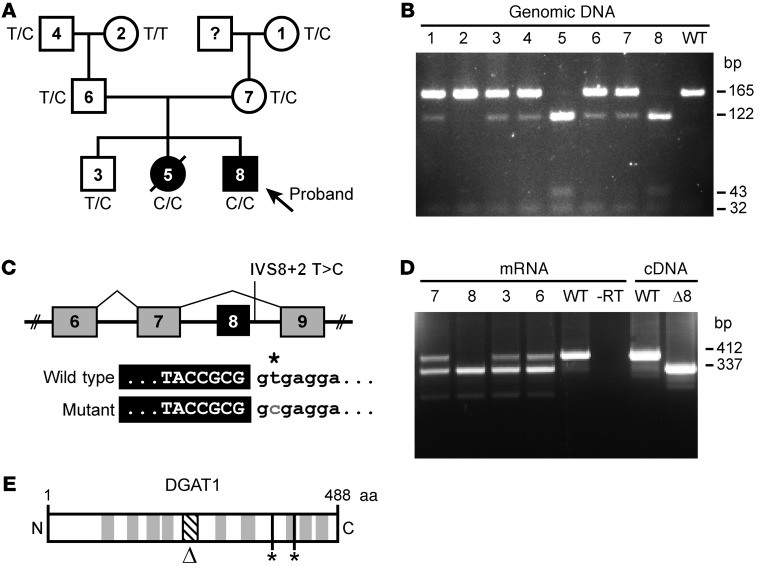
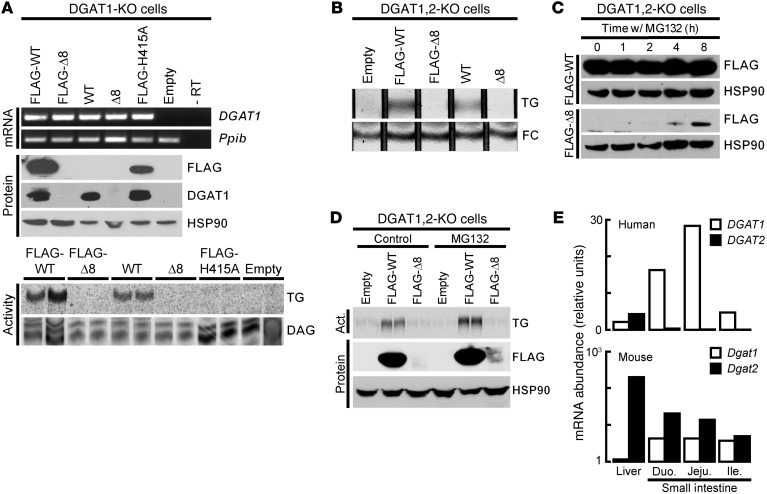
Congenital diarrheal disorders (CDDs) are a collection of rare, heterogeneous enteropathies with early onset and often severe outcomes. Here, we report a family of Ashkenazi Jewish descent, with 2 out of 3 children affected by CDD. Both affected children presented 3 days after birth with severe, intractable diarrhea. One child died from complications at age 17 months. The second child showed marked improvement, with resolution of most symptoms at 10 to 12 months of age. Using exome sequencing, we identified a rare splice site mutation in the DGAT1 gene and found that both affected children were homozygous carriers. Molecular analysis of the mutant allele indicated a total loss of function, with no detectable DGAT1 protein or activity produced. The precise cause of diarrhea is unknown, but we speculate that it relates to abnormal fat absorption and buildup of DGAT substrates in the intestinal mucosa. Our results identify DGAT1 loss-of-function mutations as a rare cause of CDDs. These findings prompt concern for DGAT1 inhibition in humans, which is being assessed for treating metabolic and other diseases.
Figures
References
-
- Berni Canani R, Terrin G, Cardillo G, Tomaiuolo R, Castaldo G. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr. 2010;50(4):360–366. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
