

Non-gaussian distributions affect identification of expression patterns, functional annotation, and prospective classification in human cancer genomes
- PMID: 23118863
- PMCID: PMC3485292
- DOI: 10.1371/journal.pone.0046935
Non-gaussian distributions affect identification of expression patterns, functional annotation, and prospective classification in human cancer genomes
Abstract
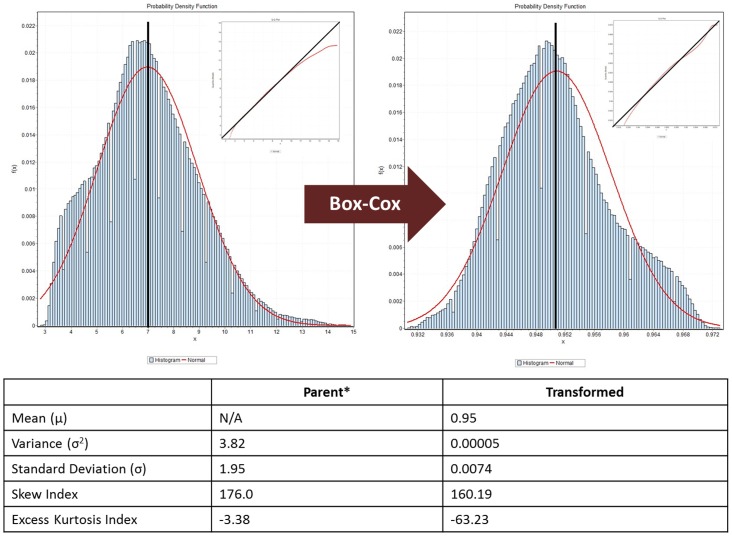
Introduction: Gene expression data is often assumed to be normally-distributed, but this assumption has not been tested rigorously. We investigate the distribution of expression data in human cancer genomes and study the implications of deviations from the normal distribution for translational molecular oncology research.
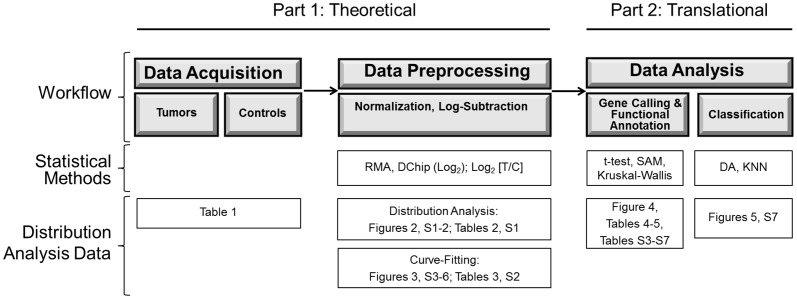
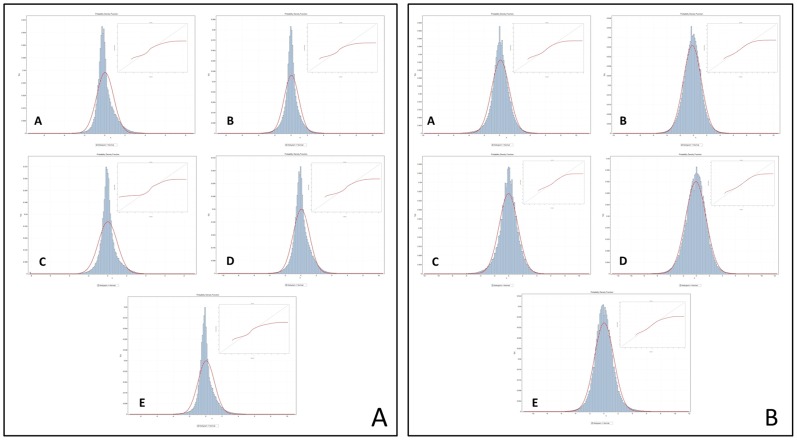
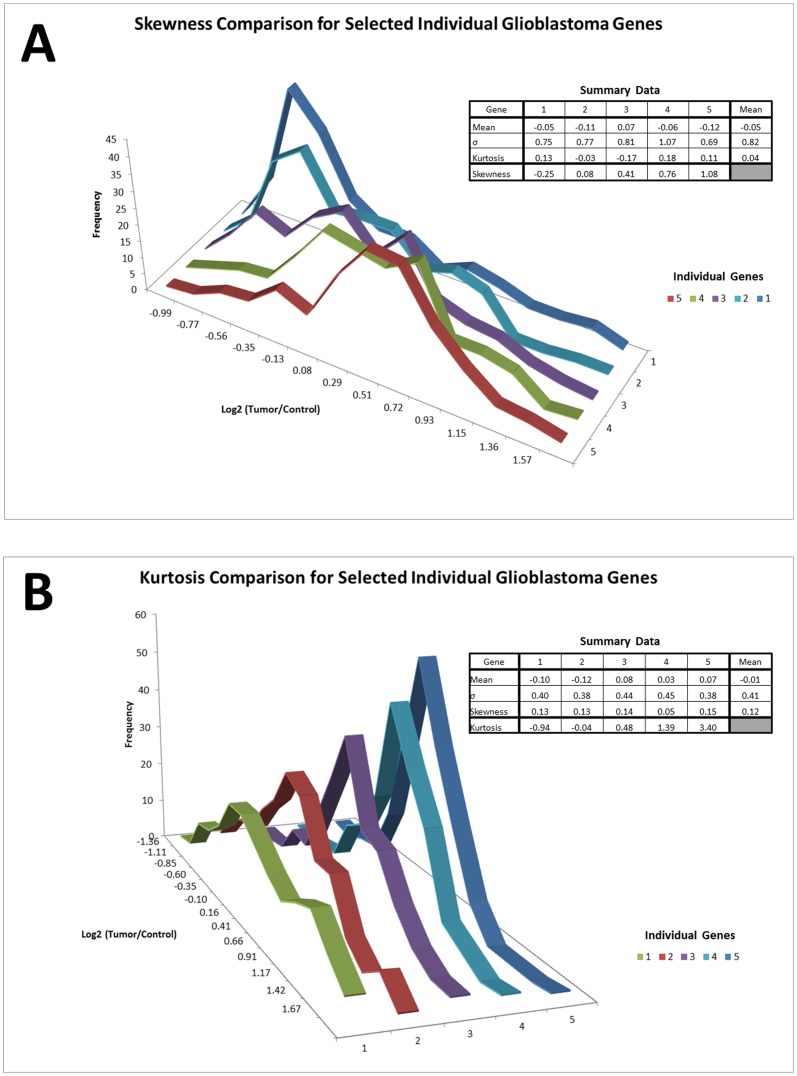
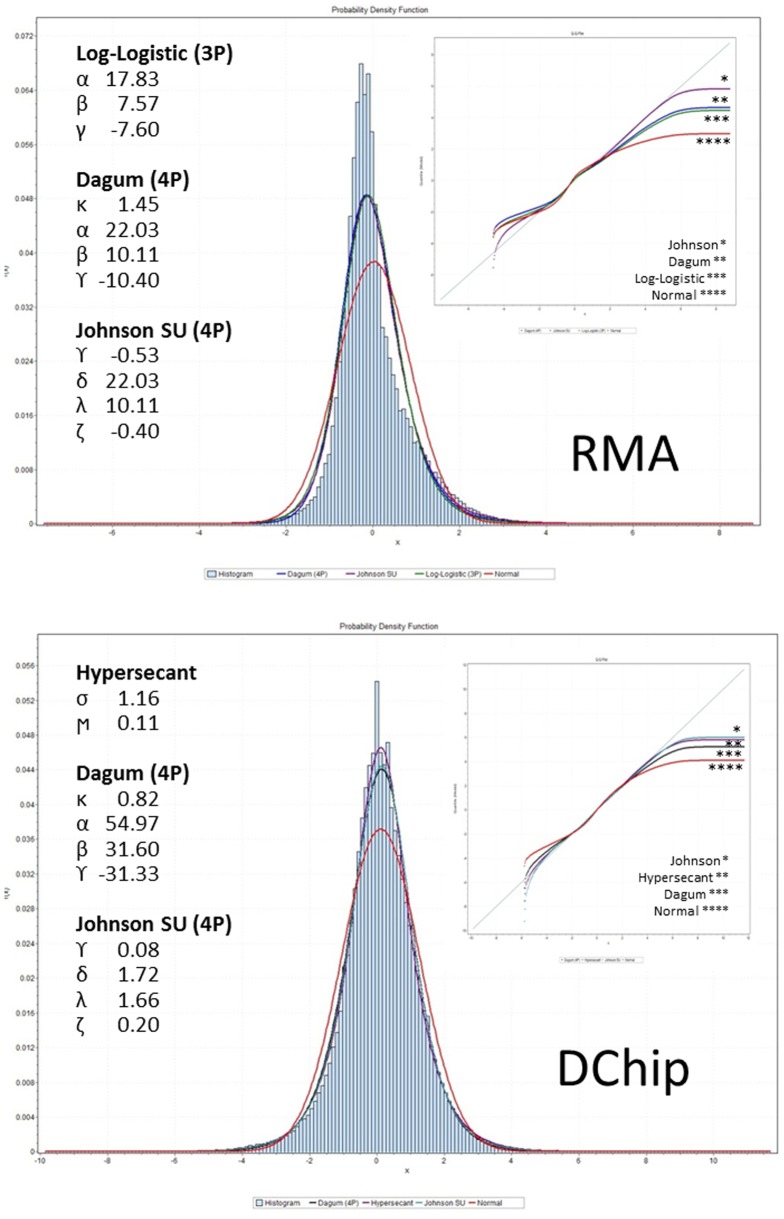
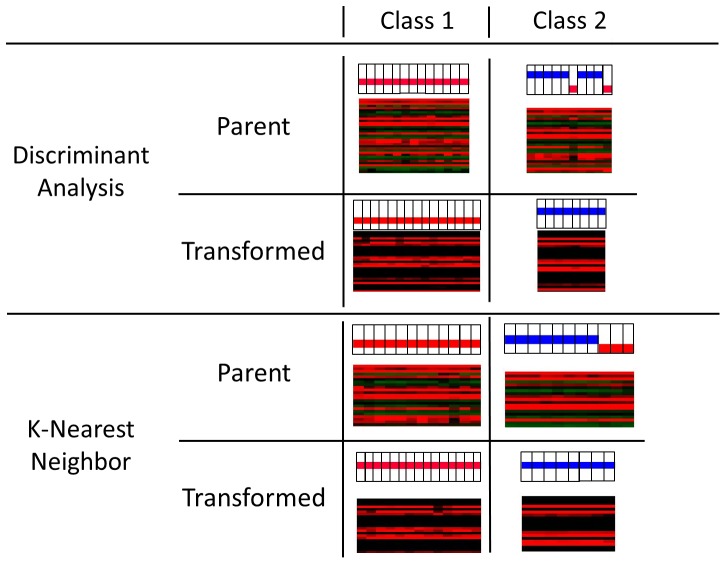
Methods: We conducted a central moments analysis of five cancer genomes and performed empiric distribution fitting to examine the true distribution of expression data both on the complete-experiment and on the individual-gene levels. We used a variety of parametric and nonparametric methods to test the effects of deviations from normality on gene calling, functional annotation, and prospective molecular classification using a sixth cancer genome.
Results: Central moments analyses reveal statistically-significant deviations from normality in all of the analyzed cancer genomes. We observe as much as 37% variability in gene calling, 39% variability in functional annotation, and 30% variability in prospective, molecular tumor subclassification associated with this effect.
Conclusions: Cancer gene expression profiles are not normally-distributed, either on the complete-experiment or on the individual-gene level. Instead, they exhibit complex, heavy-tailed distributions characterized by statistically-significant skewness and kurtosis. The non-Gaussian distribution of this data affects identification of differentially-expressed genes, functional annotation, and prospective molecular classification. These effects may be reduced in some circumstances, although not completely eliminated, by using nonparametric analytics. This analysis highlights two unreliable assumptions of translational cancer gene expression analysis: that "small" departures from normality in the expression data distributions are analytically-insignificant and that "robust" gene-calling algorithms can fully compensate for these effects.
Conflict of interest statement
Figures
References
-
- Seo J, Gordish-Dressman H, Hoffman EP (2006) An interactive power analysis tool for microarray hypothesis testing and generation. Bioinformatics 22: 808–814. - PubMed
-
- Bogner V, Leidel BA, Kanz KG, Mutschler W, Neugebauer EA, et al. (2011) Pathway analysis in microarray data: a comparison of two different pathway analysis devices in the same data set. Shock 35: 245–251. - PubMed
-
- Hardiman G (2008) Applications of microarrays and biochips in pharmacogenomics. Methods Mol Biol 448: 21–30. - PubMed
-
- Marko NF, Prayson RA, Barnett GH, Weil RJ (2010) Integrated molecular analysis suggests a three-class model for low-grade gliomas: a proof-of-concept study. Genomics 95: 16–24. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
