

Neuroprotective actions of methylene blue and its derivatives
- PMID: 23118969
- PMCID: PMC3485214
- DOI: 10.1371/journal.pone.0048279
Neuroprotective actions of methylene blue and its derivatives
Abstract
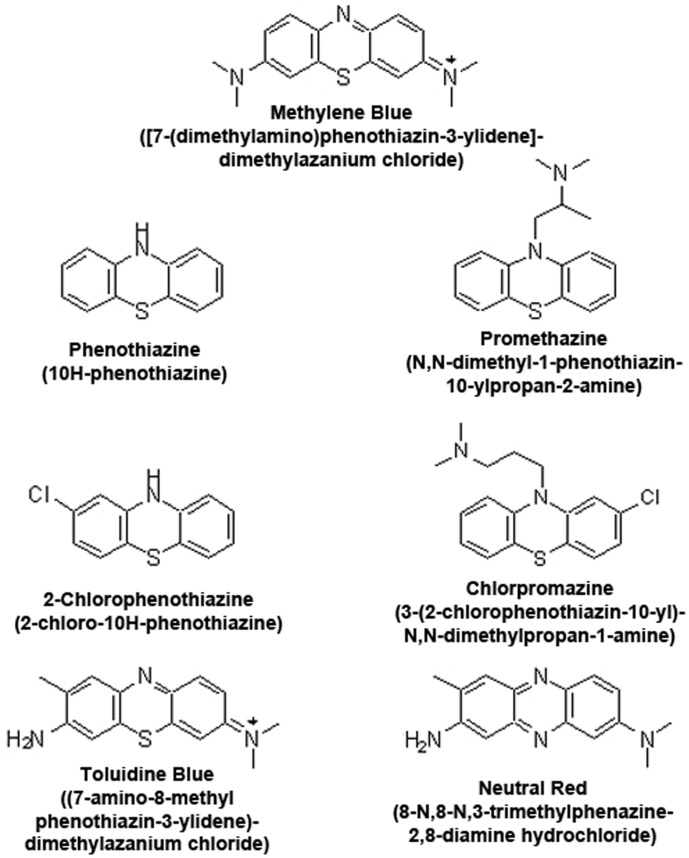
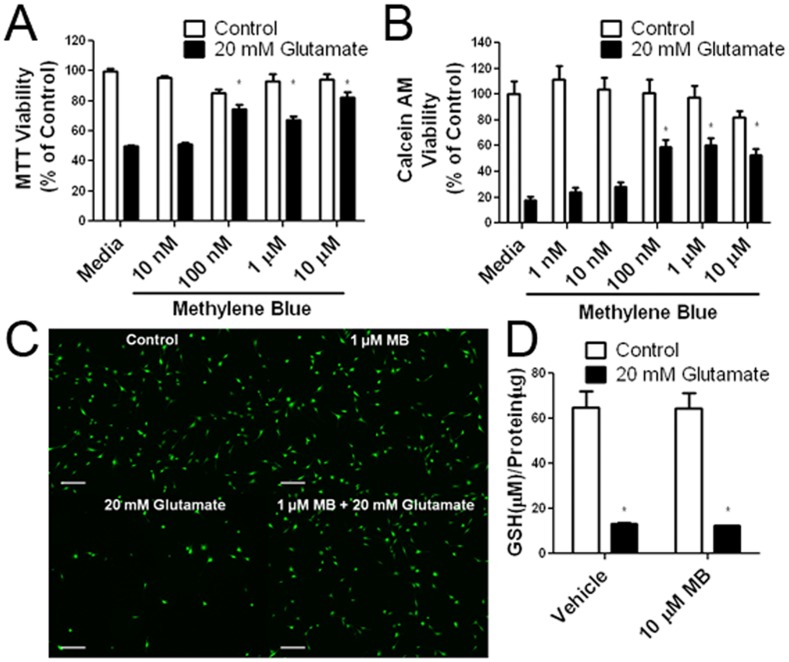
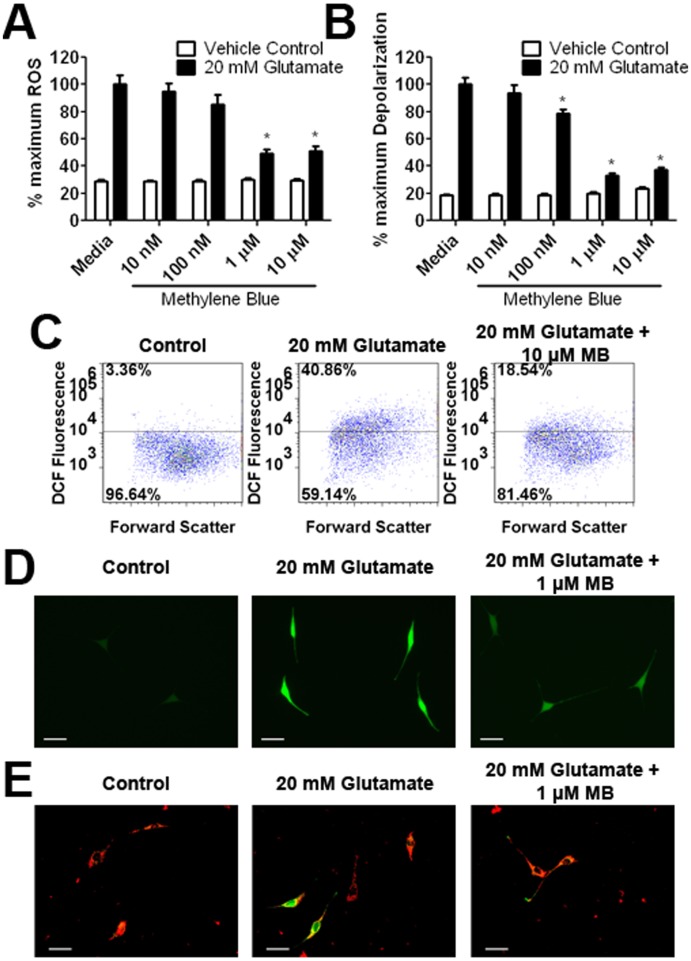
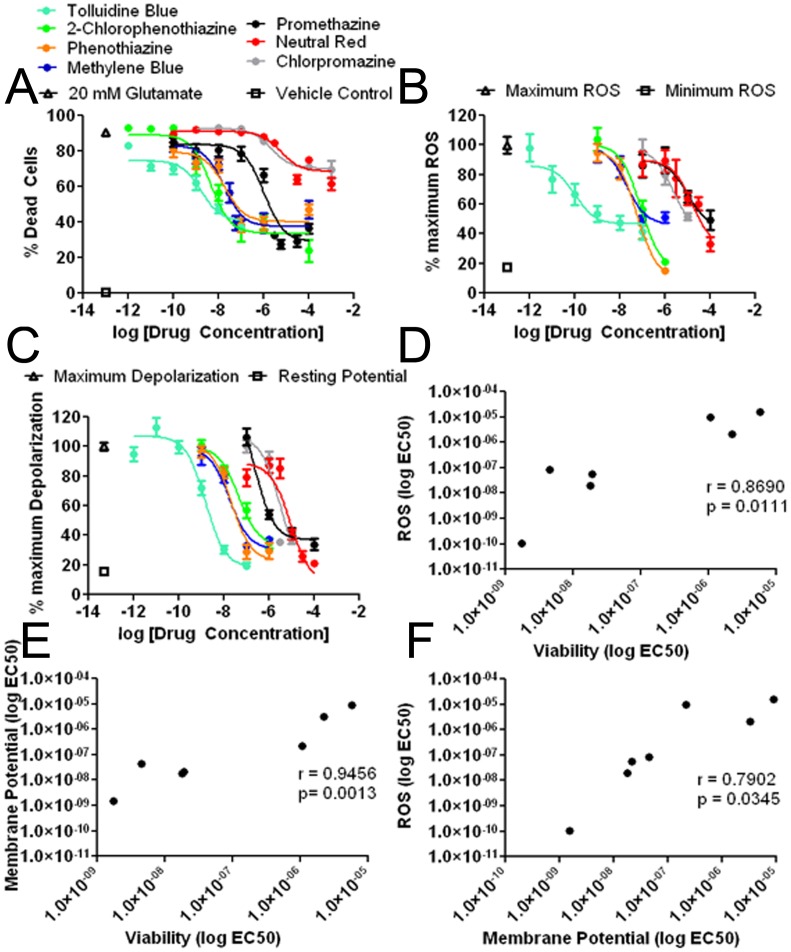
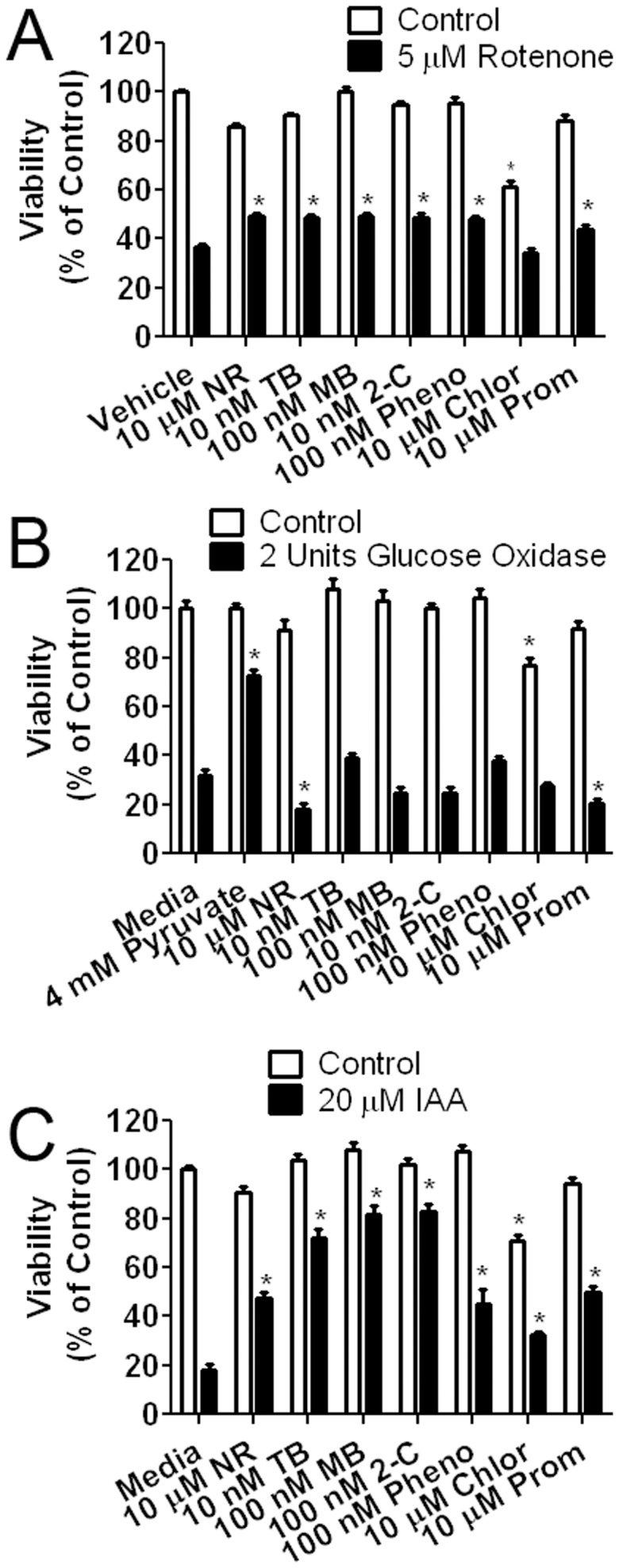
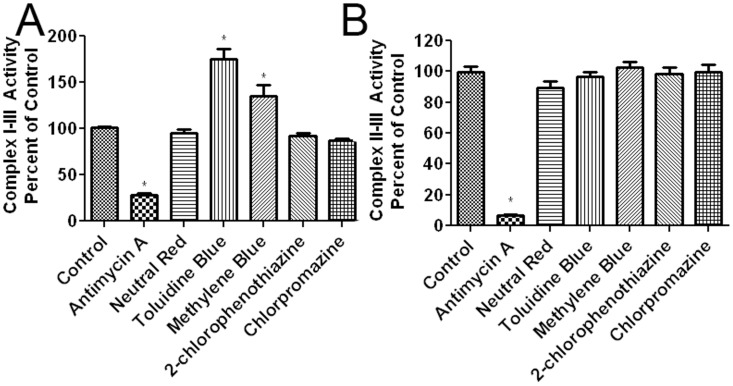
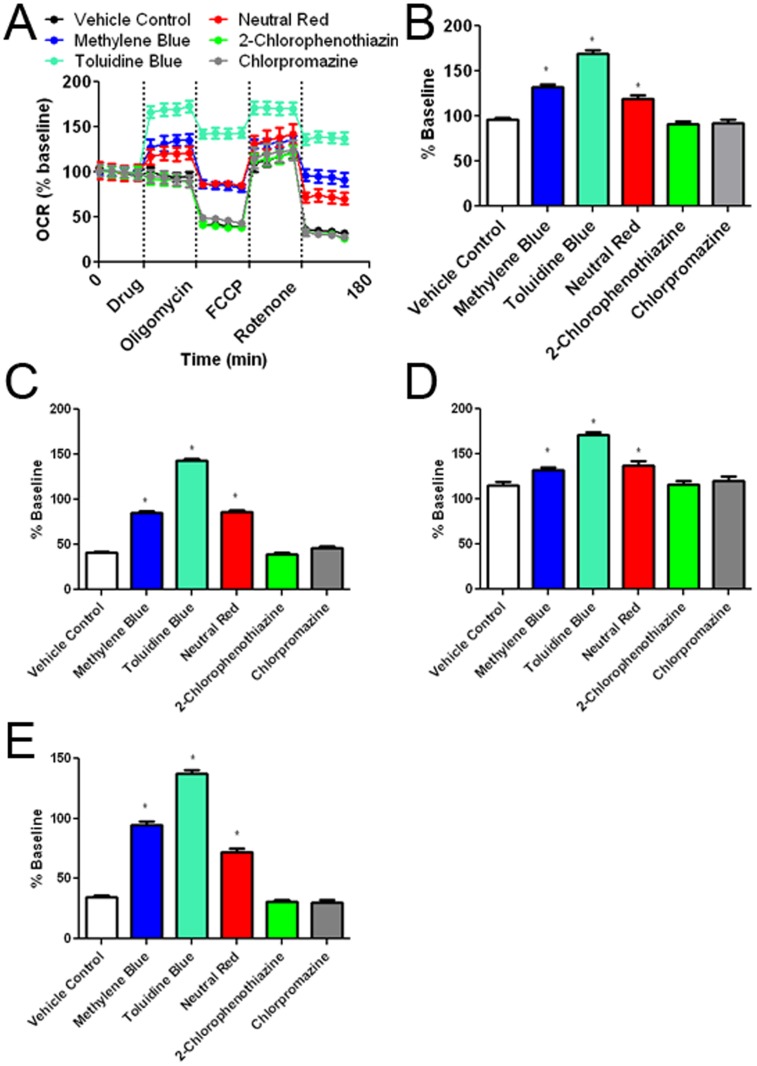
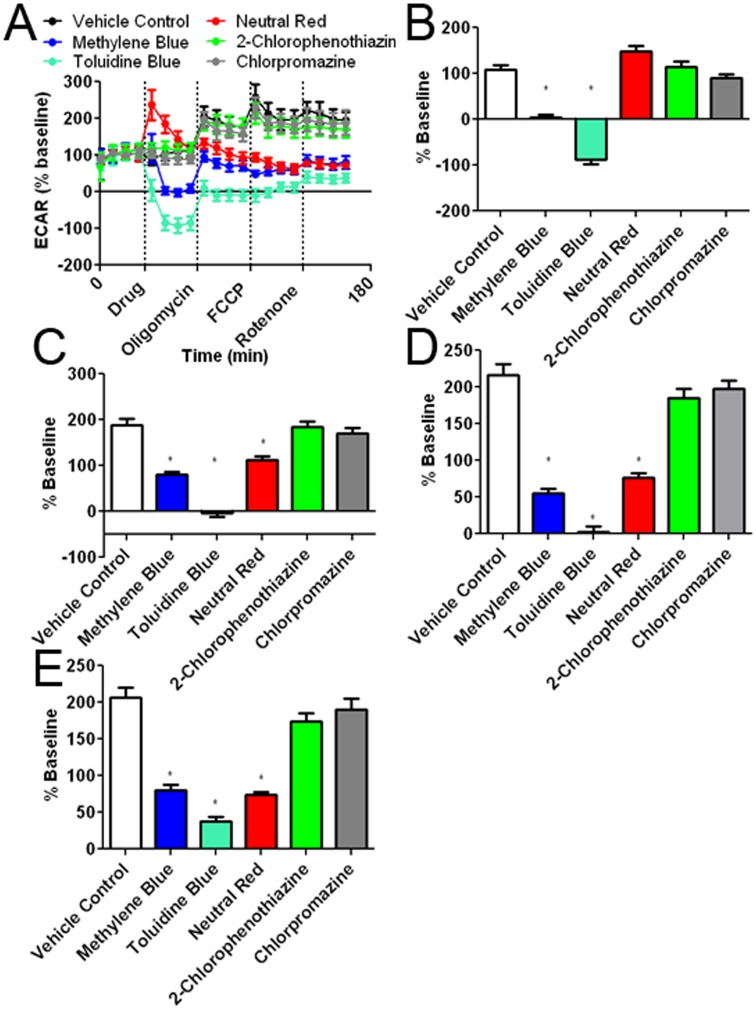
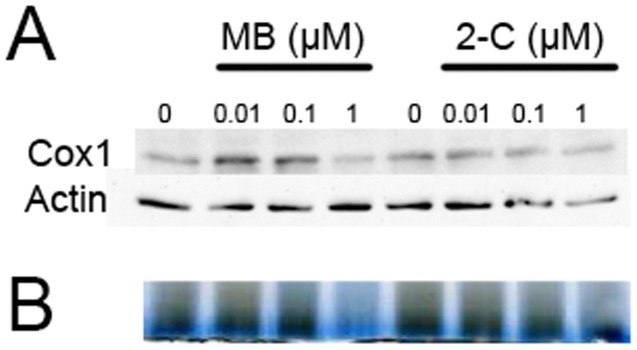
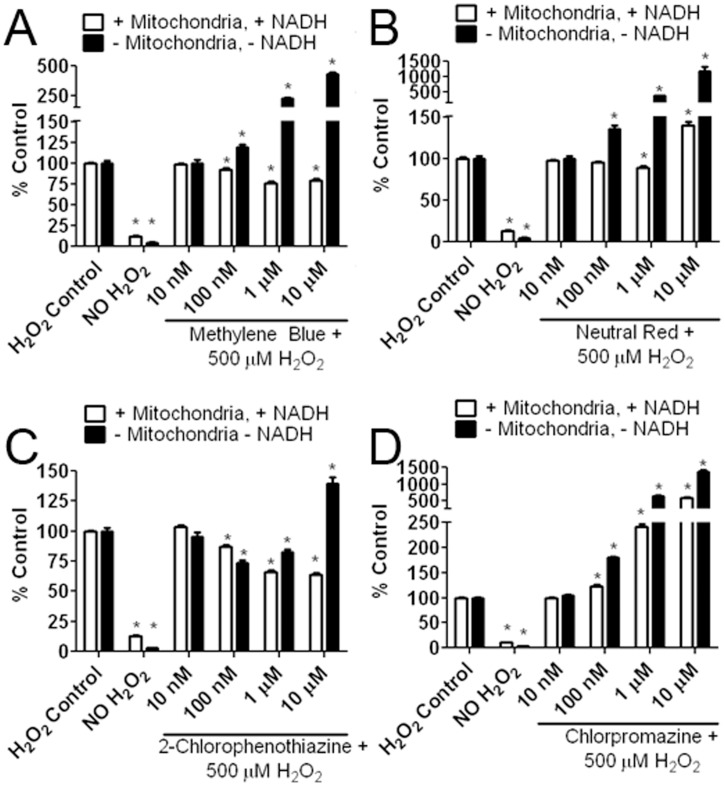
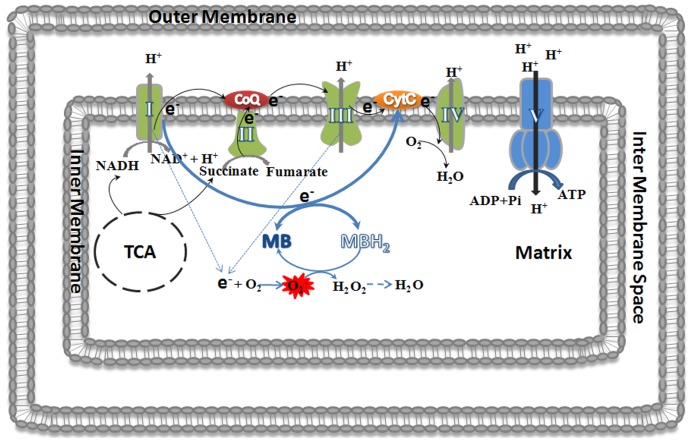
Methylene blue (MB), the first lead chemical structure of phenothiazine and other derivatives, is commonly used in diagnostic procedures and as a treatment for methemoglobinemia. We have previously demonstrated that MB could function as an alternative mitochondrial electron transfer carrier, enhance cellular oxygen consumption, and provide protection in vitro and in rodent models of Parkinson's disease and stroke. In the present study, we investigated the structure-activity relationships of MB in vitro using MB and six structurally related compounds. MB reduces mitochondrial superoxide production via alternative electron transfer that bypasses mitochondrial complexes I-III. MB mitigates reactive free radical production and provides neuroprotection in HT-22 cells against glutamate, IAA and rotenone toxicity. Distinctly, MB provides no protection against direct oxidative stress induced by glucose oxidase. Substitution of a side chain at MB's 10-nitrogen rendered a 1000-fold reduction of the protective potency against glutamate neurototoxicity. Compounds without side chains at positions 3 and 7, chlorophenothiazine and phenothiazine, have distinct redox potentials compared to MB and are incapable of enhancing mitochondrial electron transfer, while obtaining direct antioxidant actions against glutamate, IAA, and rotenone insults. Chlorophenothiazine exhibited direct antioxidant actions in mitochondria lysate assay compared to MB, which required reduction by NADH and mitochondria. MB increased complex IV expression and activity, while 2-chlorphenothiazine had no effect. Our study indicated that MB could attenuate superoxide production by functioning as an alternative mitochondrial electron transfer carrier and as a regenerable anti-oxidant in mitochondria.
Conflict of interest statement
Figures
References
-
- Bergen DC (2008) Neurological Disorders: Public Health Challenges. Arch Neurol 65: 154–154.
-
- Pratico D (2008) Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci 29: 609–615. - PubMed
-
- Barnham KJ, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3: 205–214. - PubMed
-
- Sanchez-Moreno C, Dashe JF, Scott T, Thaler D, Folstein MF, et al. (2004) Decreased levels of plasma vitamin C and increased concentrations of inflammatory and oxidative stress markers after stroke. Stroke 35: 163–168. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
