

Defective complement inhibitory function predisposes to renal disease
- PMID: 23121180
- PMCID: PMC3941008
- DOI: 10.1146/annurev-med-072211-110606
Defective complement inhibitory function predisposes to renal disease
Abstract
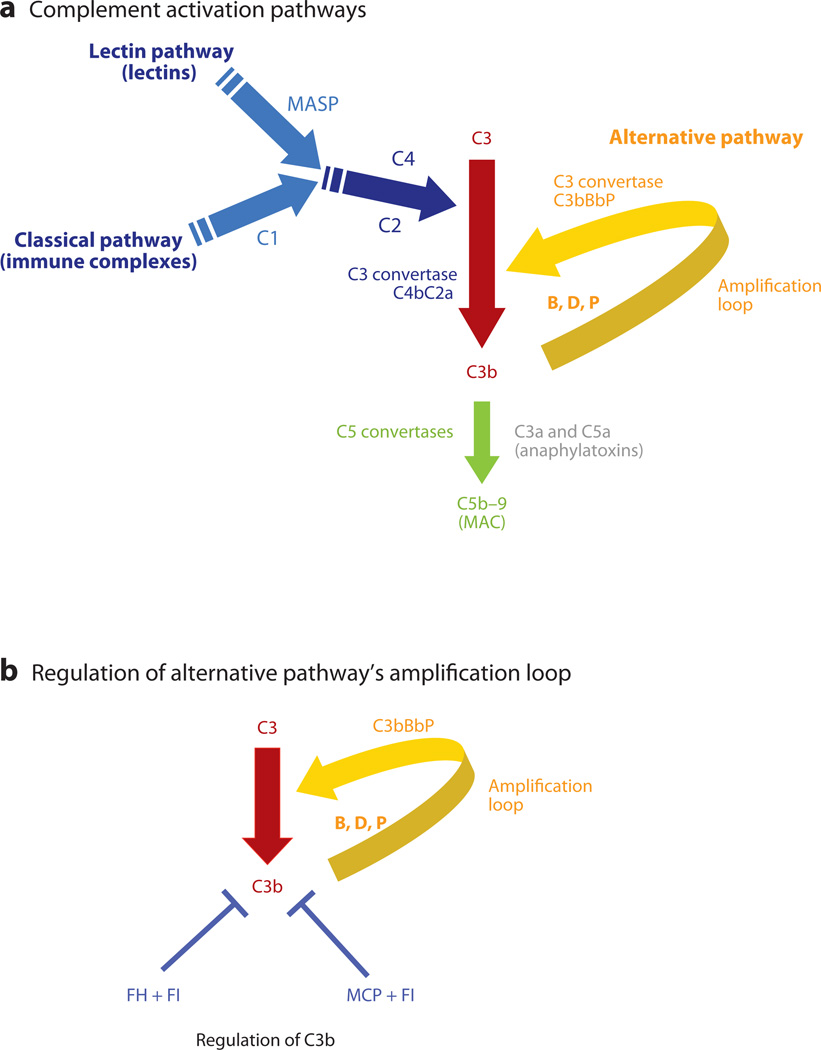
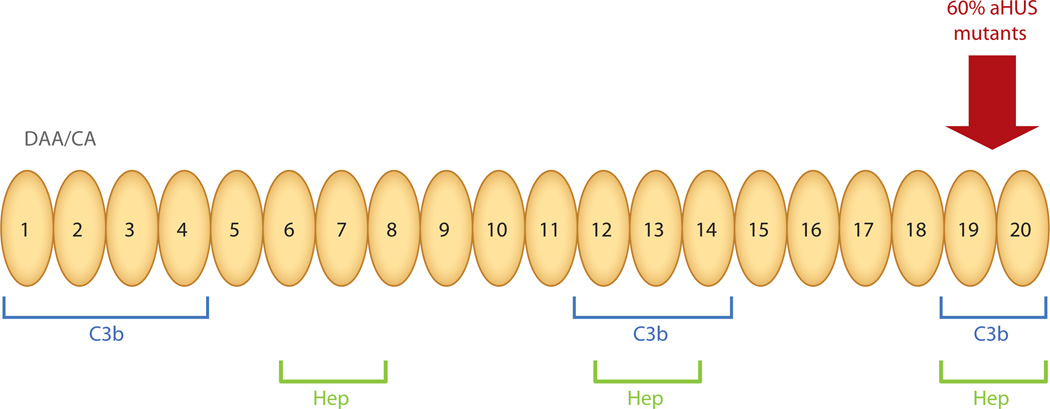
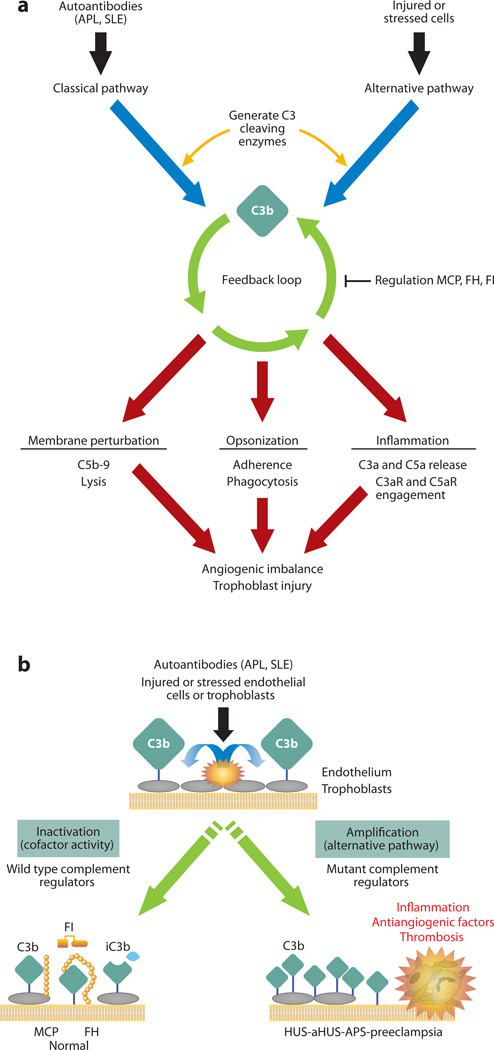
The role of the complement system in mediating human renal disease has long been recognized in immune-complex excess syndromes such as systemic lupus erythematosus and in dense deposit disease in which no immunoglobulin (Ig) is present. Over the past 15 years, mutations in complement regulatory genes have been demonstrated to predispose to thrombotic microangiopathies including atypical hemolytic uremic syndrome, C3 and C1q glomerulopathies, and preeclampsia. Excessive complement activation on an endothelial cell, due to either an autoantibody or a regulatory protein deficiency, sets up a procoagulant state in these diseases as well as in the antiphospholipid syndrome. Knowledge of the genes involved and the functional consequences of alterations in their structure has led to therapy that blocks complement activation.
Figures
References
-
- Markiewski MM, Nilsson B, Ekdahl KN, et al. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28:184–192. - PubMed
-
-
Richards A, Kavanagh D, Atkinson JP. Inherited complement regulatory protein deficiency predisposes to human disease in acute injury and chronic inflammatory states: the examples of vascular damage in atypical hemolytic uremic syndrome and debris accumulation in age-related macular degeneration. Adv. Immunol. 2007;96:141–177. Recent, comprehensive, and authoritative reviews on aHUS.
-
-
- Abarrategui-Garrido C, Martinez-Barricarte R, Lopez-Trascasa M, et al. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations ofCFHR1 associated with atypical hemolytic uremic syndrome. Blood. 2009;114:4261–4271. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
