

Mutations of the gene encoding otogelin are a cause of autosomal-recessive nonsyndromic moderate hearing impairment
- PMID: 23122587
- PMCID: PMC3487128
- DOI: 10.1016/j.ajhg.2012.09.012
Mutations of the gene encoding otogelin are a cause of autosomal-recessive nonsyndromic moderate hearing impairment
Abstract
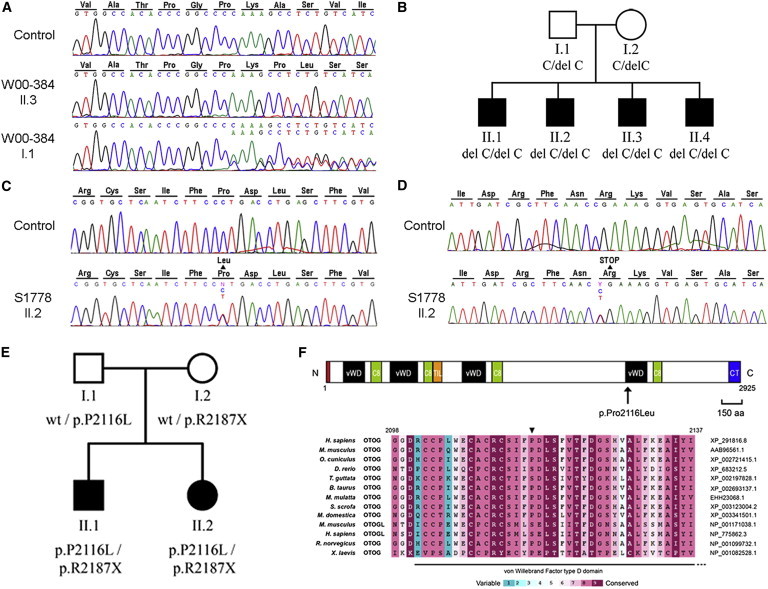
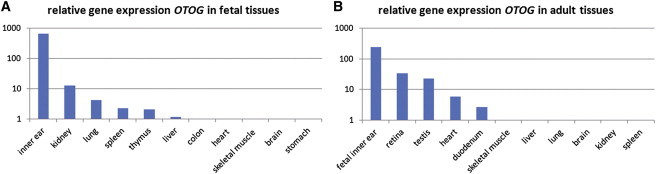
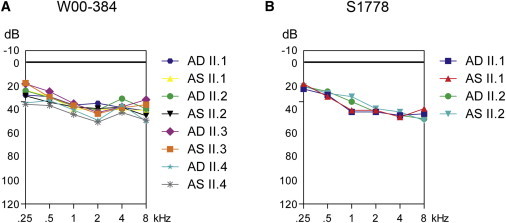
Already 40 genes have been identified for autosomal-recessive nonsyndromic hearing impairment (arNSHI); however, many more genes are still to be identified. In a Dutch family segregating arNSHI, homozygosity mapping revealed a 2.4 Mb homozygous region on chromosome 11 in p15.1-15.2, which partially overlapped with the previously described DFNB18 locus. However, no putative pathogenic variants were found in USH1C, the gene mutated in DFNB18 hearing impairment. The homozygous region contained 12 additional annotated genes including OTOG, the gene encoding otogelin, a component of the tectorial membrane. It is thought that otogelin contributes to the stability and strength of this membrane through interaction or stabilization of its constituent fibers. The murine orthologous gene was already known to cause hearing loss when defective. Analysis of OTOG in the Dutch family revealed a homozygous 1 bp deletion, c.5508delC, which leads to a shift in the reading frame and a premature stop codon, p.Ala1838ProfsX31. Further screening of 60 unrelated probands from Spanish arNSHI families detected compound heterozygous OTOG mutations in one family, c.6347C>T (p.Pro2116Leu) and c. 6559C>T (p.Arg2187X). The missense mutation p.Pro2116Leu affects a highly conserved residue in the fourth von Willebrand factor type D domain of otogelin. The subjects with OTOG mutations have a moderate hearing impairment, which can be associated with vestibular dysfunction. The flat to shallow "U" or slightly downsloping shaped audiograms closely resembled audiograms of individuals with recessive mutations in the gene encoding α-tectorin, another component of the tectorial membrane. This distinctive phenotype may represent a clue to orientate the molecular diagnosis.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Lenz D.R., Avraham K.B. Hereditary hearing loss: from human mutation to mechanism. Hear. Res. 2011;281:3–10. - PubMed
-
- Brown S.D., Hardisty-Hughes R.E., Mburu P. Quiet as a mouse: dissecting the molecular and genetic basis of hearing. Nat. Rev. Genet. 2008;9:277–290. - PubMed
-
- Dror A.A., Avraham K.B. Hearing impairment: a panoply of genes and functions. Neuron. 2010;68:293–308. - PubMed
-
- Goodyear R.J., Legan P.K., Wright M.B., Marcotti W., Oganesian A., Coats S.A., Booth C.J., Kros C.J., Seifert R.A., Bowen-Pope D.F., Richardson G.P. A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J. Neurosci. 2003;23:9208–9219. - PMC - PubMed
-
- Grillet N., Schwander M., Hildebrand M.S., Sczaniecka A., Kolatkar A., Velasco J., Webster J.A., Kahrizi K., Najmabadi H., Kimberling W.J. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am. J. Hum. Genet. 2009;85:328–337. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
