

DHEA metabolites activate estrogen receptors alpha and beta
- PMID: 23123738
- PMCID: PMC3529809
- DOI: 10.1016/j.steroids.2012.10.002
DHEA metabolites activate estrogen receptors alpha and beta
Abstract
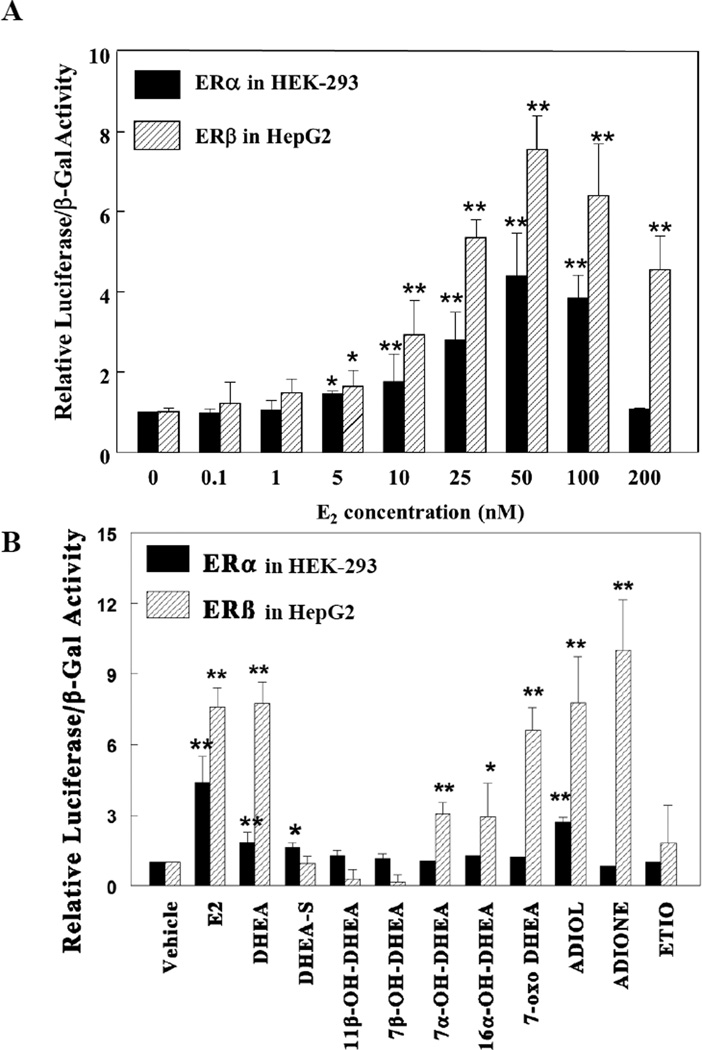
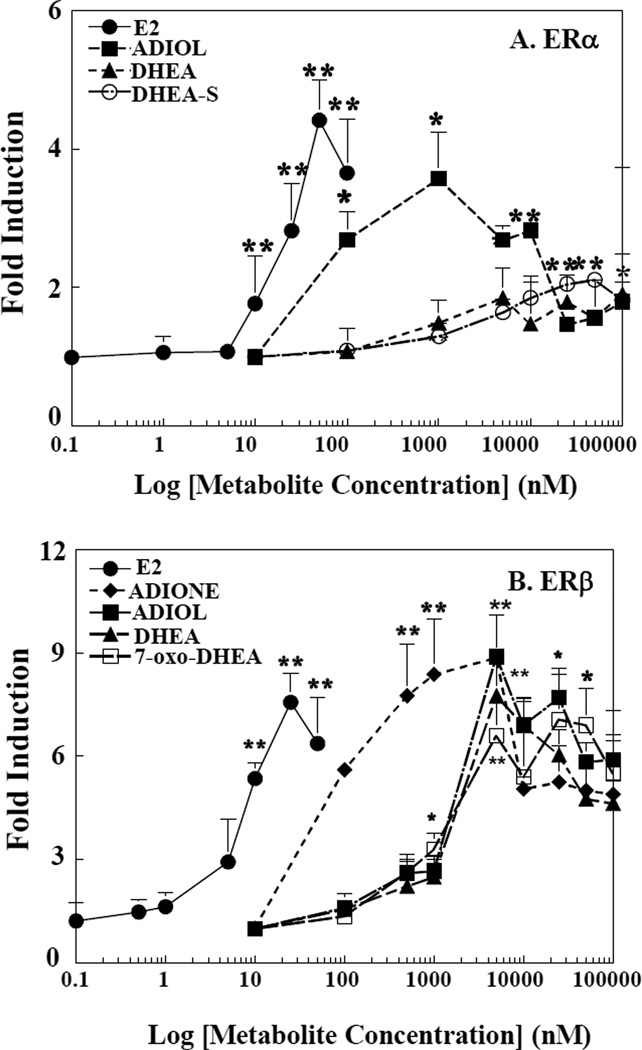
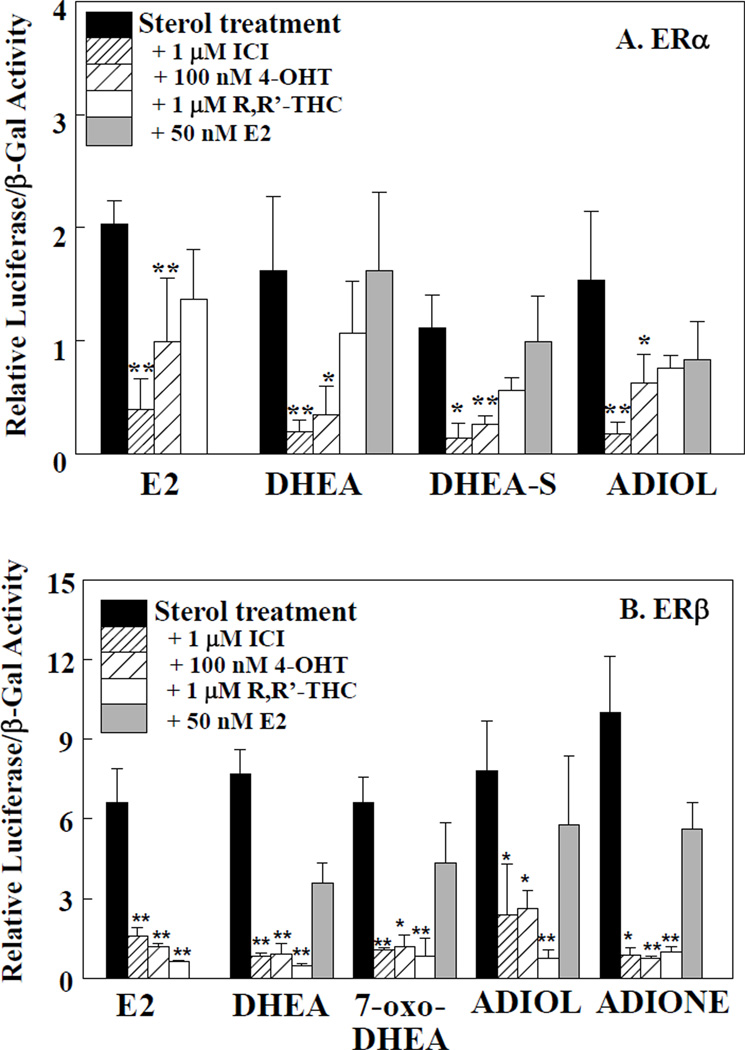
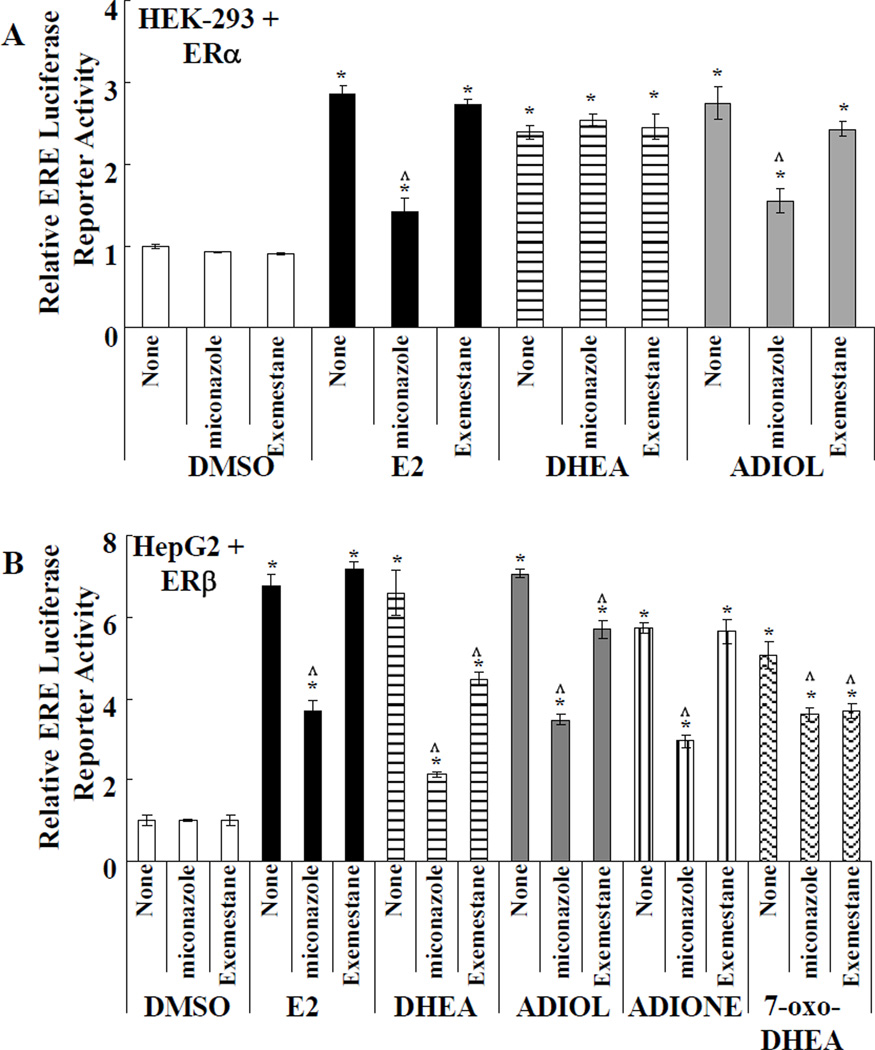
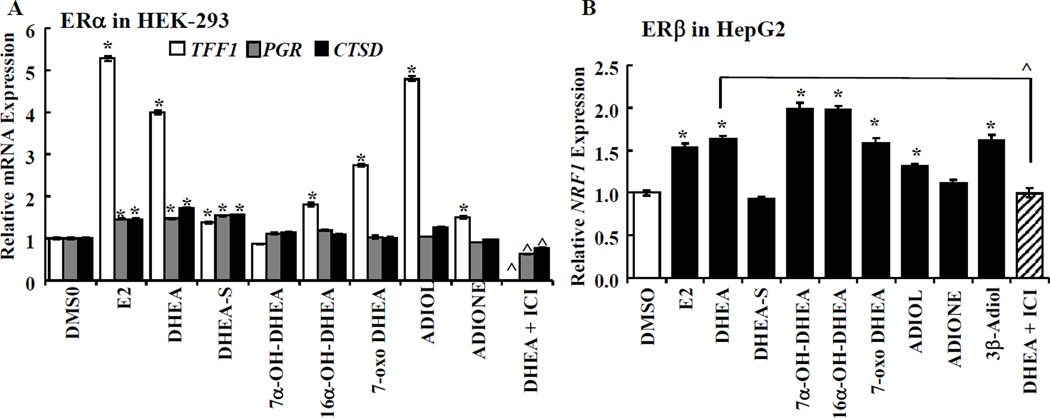
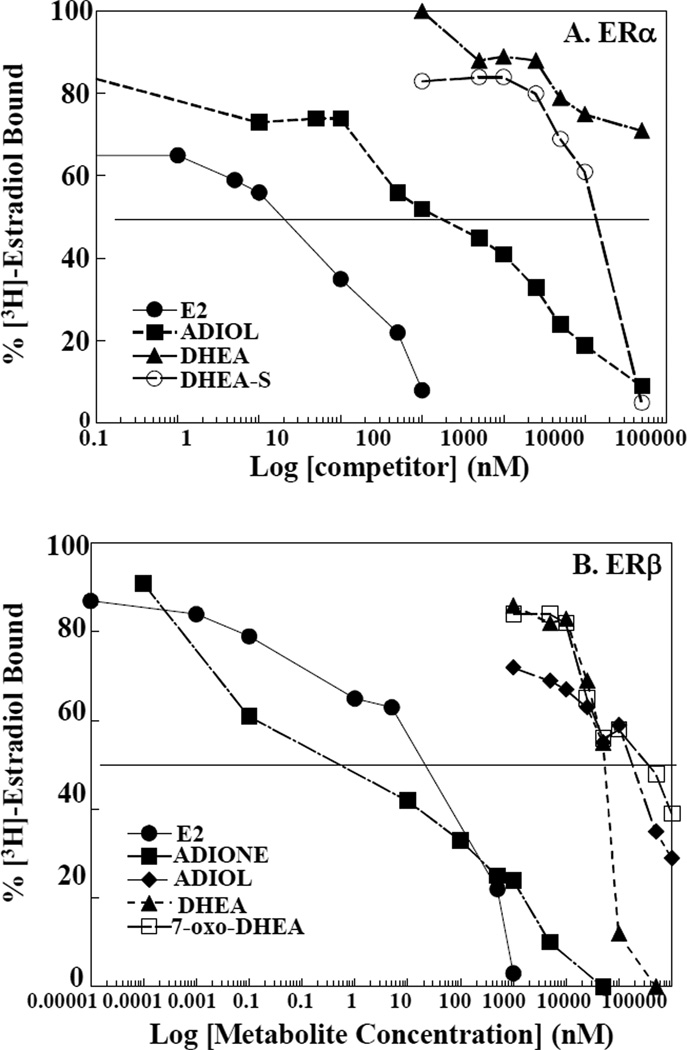
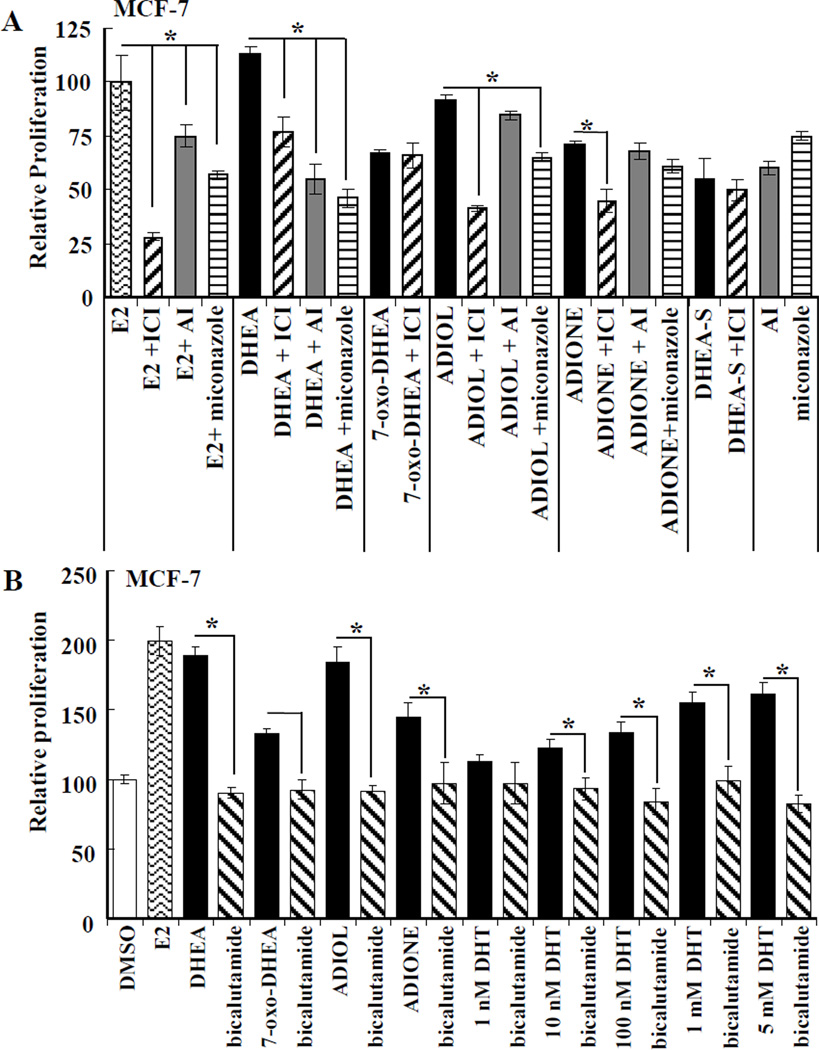
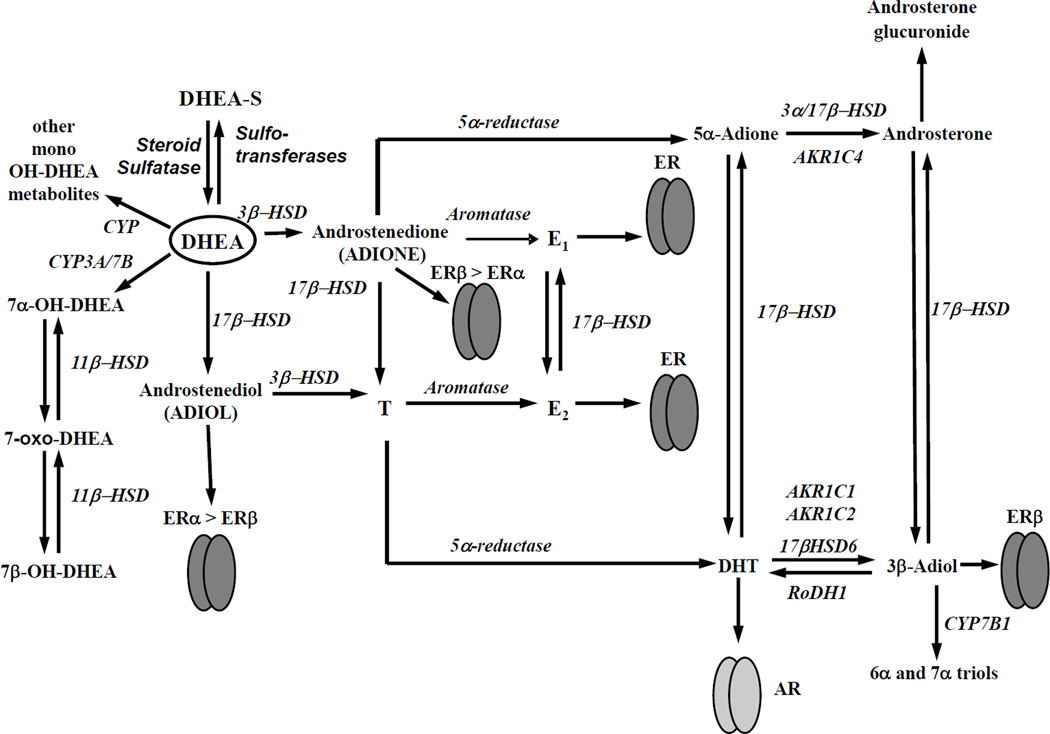
Dehydroepiandrosterone (DHEA) levels were reported to associate with increased breast cancer risk in postmenopausal women, but some carcinogen-induced rat mammary tumor studies question this claim. The purpose of this study was to determine how DHEA and its metabolites affect estrogen receptors α or β (ERα or ERβ)-regulated gene transcription and cell proliferation. In transiently transfected HEK-293 cells, androstenediol, DHEA, and DHEA-S activated ERα. In ERβ transfected HepG2 cells, androstenedione, DHEA, androstenediol, and 7-oxo DHEA stimulated reporter activity. ER antagonists ICI 182,780 (fulvestrant) and 4-hydroxytamoxifen, general P450 inhibitor miconazole, and aromatase inhibitor exemestane inhibited activation by DHEA or metabolites in transfected cells. ERβ-selective antagonist R,R-THC (R,R-cis-diethyl tetrahydrochrysene) inhibited DHEA and DHEA metabolite transcriptional activity in ERβ-transfected cells. Expression of endogenous estrogen-regulated genes: pS2, progesterone receptor, cathepsin D1, and nuclear respiratory factor-1 was increased by DHEA and its metabolites in an ER-subtype, gene, and cell-specific manner. DHEA metabolites, but not DHEA, competed with 17β-estradiol for ERα and ERβ binding and stimulated MCF-7 cell proliferation, demonstrating that DHEA metabolites interact directly with ERα and ERβin vitro, modulating estrogen target genes in vivo.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Simpson ER, Davis SR. Minireview: aromatase and the regulation of estrogen biosynthesis-some new perspectives. Endocrinology. 2001;142:4589–4594. - PubMed
-
- Janni W, Hepp P. Adjuvant aromatase inhibitor therapy: Outcomes and safety. Cancer Treat Rev. 2010;36:249–261. - PubMed
-
- Kaaks R, Rinaldi S, Key TJ, Berrino F, Peeters PHM, Biessy C, et al. Postmenopausal serum androgens, oestrogens and breast cancer risk: the European prospective investigation into cancer and nutrition. Endocr Relat Cancer. 2005;12:1071–1082. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
