

Efficient assembly and annotation of the transcriptome of catfish by RNA-Seq analysis of a doubled haploid homozygote
- PMID: 23127152
- PMCID: PMC3582483
- DOI: 10.1186/1471-2164-13-595
Efficient assembly and annotation of the transcriptome of catfish by RNA-Seq analysis of a doubled haploid homozygote
Abstract
Background: Upon the completion of whole genome sequencing, thorough genome annotation that associates genome sequences with biological meanings is essential. Genome annotation depends on the availability of transcript information as well as orthology information. In teleost fish, genome annotation is seriously hindered by genome duplication. Because of gene duplications, one cannot establish orthologies simply by homology comparisons. Rather intense phylogenetic analysis or structural analysis of orthologies is required for the identification of genes. To conduct phylogenetic analysis and orthology analysis, full-length transcripts are essential. Generation of large numbers of full-length transcripts using traditional transcript sequencing is very difficult and extremely costly.
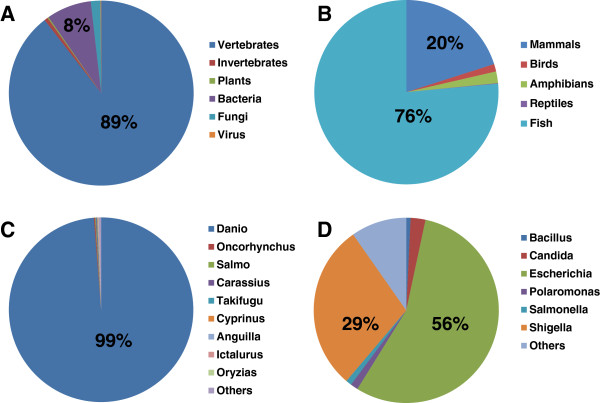
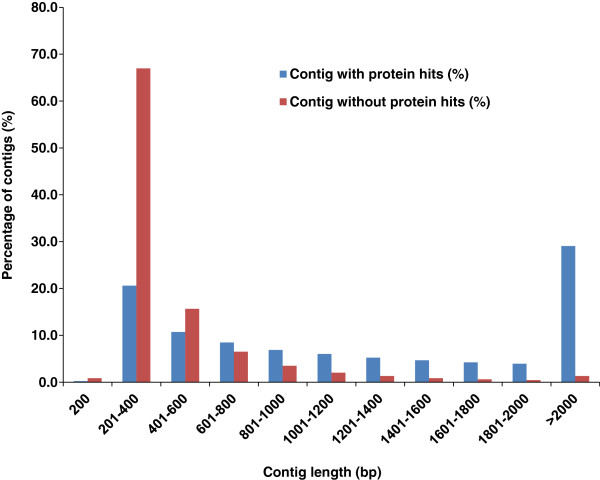
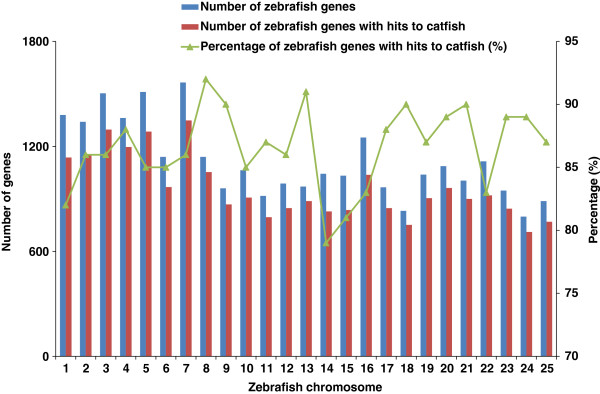
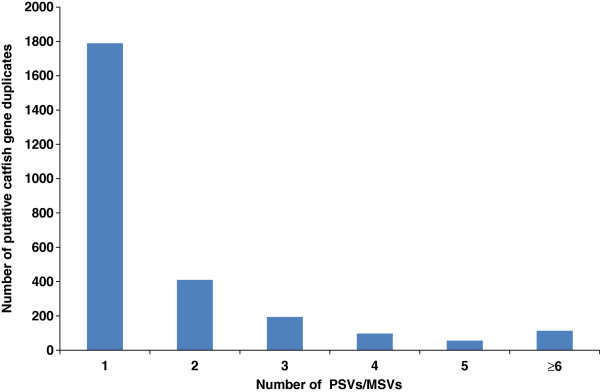
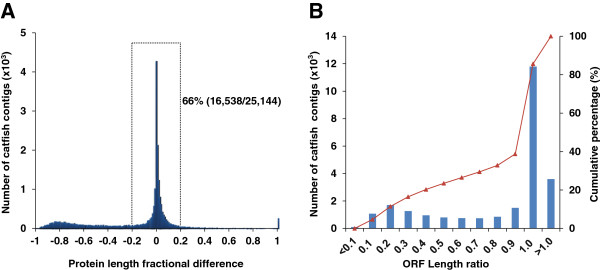
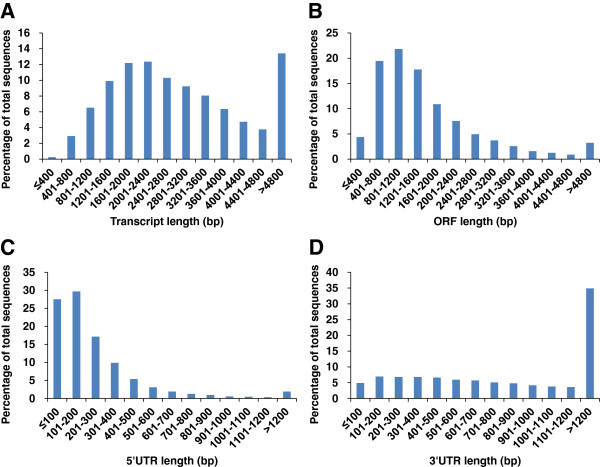
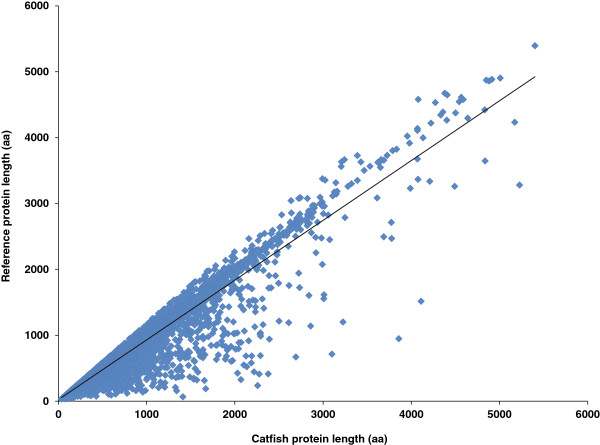
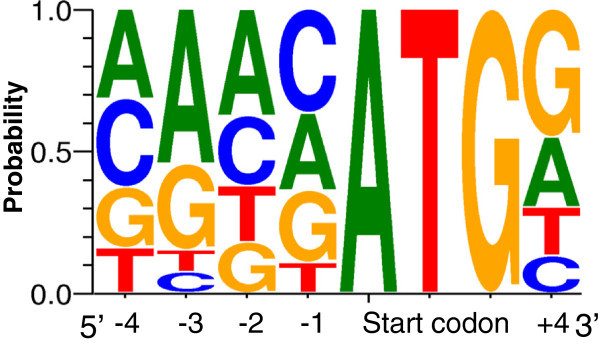
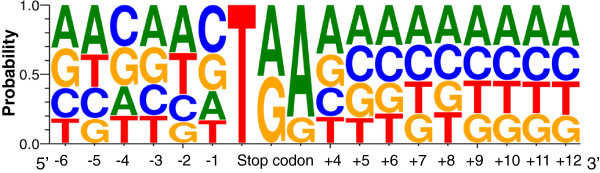
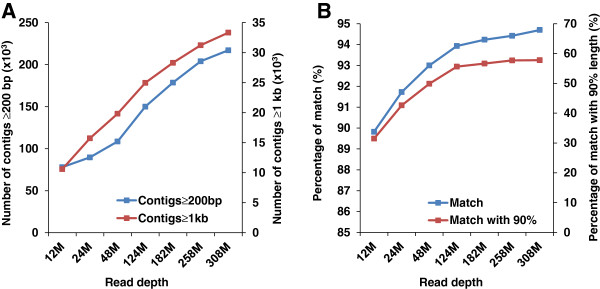
Results: In this work, we took advantage of a doubled haploid catfish, which has two sets of identical chromosomes and in theory there should be no allelic variations. As such, transcript sequences generated from next-generation sequencing can be favorably assembled into full-length transcripts. Deep sequencing of the doubled haploid channel catfish transcriptome was performed using Illumina HiSeq 2000 platform, yielding over 300 million high-quality trimmed reads totaling 27 Gbp. Assembly of these reads generated 370,798 non-redundant transcript-derived contigs. Functional annotation of the assembly allowed identification of 25,144 unique protein-encoding genes. A total of 2,659 unique genes were identified as putative duplicated genes in the catfish genome because the assembly of the corresponding transcripts harbored PSVs or MSVs (in the form of pseudo-SNPs in the assembly). Of the 25,144 contigs with unique protein hits, around 20,000 contigs matched 50% length of reference proteins, and over 14,000 transcripts were identified as full-length with complete open reading frames. The characterization of consensus sequences surrounding start codon and the stop codon confirmed the correct assembly of the full-length transcripts.
Conclusions: The large set of transcripts assembled in this study is the most comprehensive set of genome resources ever developed from catfish, which will provide the much needed resources for functional genome research in catfish, serving as a reference transcriptome for genome annotation, analysis of gene duplication, gene family structures, and digital gene expression analysis. The putative set of duplicated genes provide a starting point for genome scale analysis of gene duplication in the catfish genome, and should be a valuable resource for comparative genome analysis, genome evolution, and genome function studies.
Figures
References
-
- Adamidi C, Wang Y, Gruen D, Mastrobuoni G, You X, Tolle D, Dodt M, Mackowiak SD, Gogol-Doering A, Oenal P. et al.De novo assembly and validation of planaria transcriptome by massive parallel sequencing and shotgun proteomics. Genome Res. 2011;21(7):1193–1200. doi: 10.1101/gr.113779.110. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
