

Arginase II Promotes Macrophage Inflammatory Responses Through Mitochondrial Reactive Oxygen Species, Contributing to Insulin Resistance and Atherogenesis
- PMID: 23130157
- PMCID: PMC3487353
- DOI: 10.1161/JAHA.112.000992
Arginase II Promotes Macrophage Inflammatory Responses Through Mitochondrial Reactive Oxygen Species, Contributing to Insulin Resistance and Atherogenesis
Abstract
Background: Macrophage-mediated chronic inflammation is mechanistically linked to insulin resistance and atherosclerosis. Although arginase I is considered antiinflammatory, the role of arginase II (Arg-II) in macrophage function remains elusive. This study characterizes the role of Arg-II in macrophage inflammatory responses and its impact on obesity-linked type II diabetes mellitus and atherosclerosis.
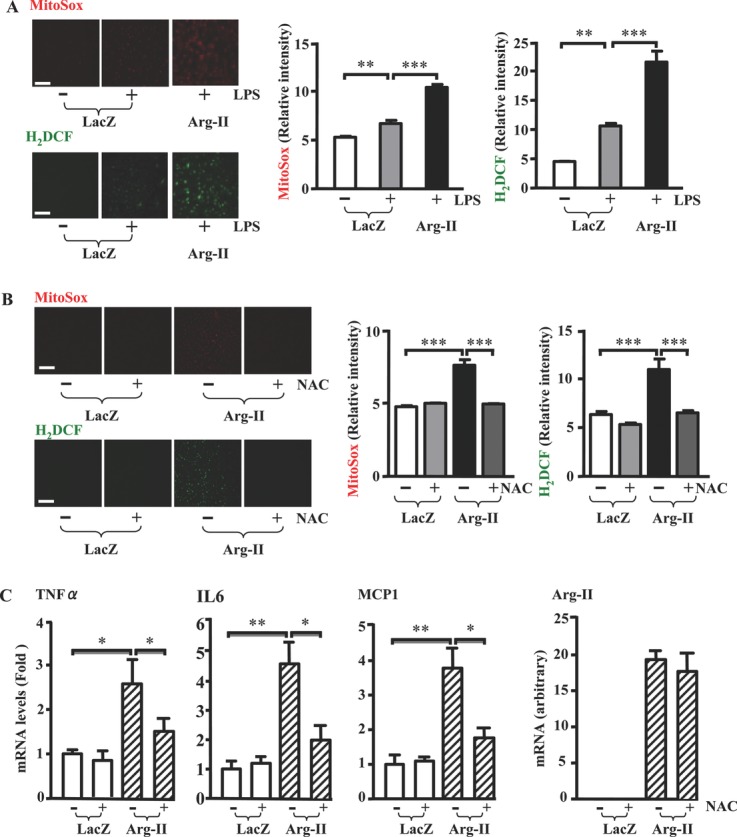
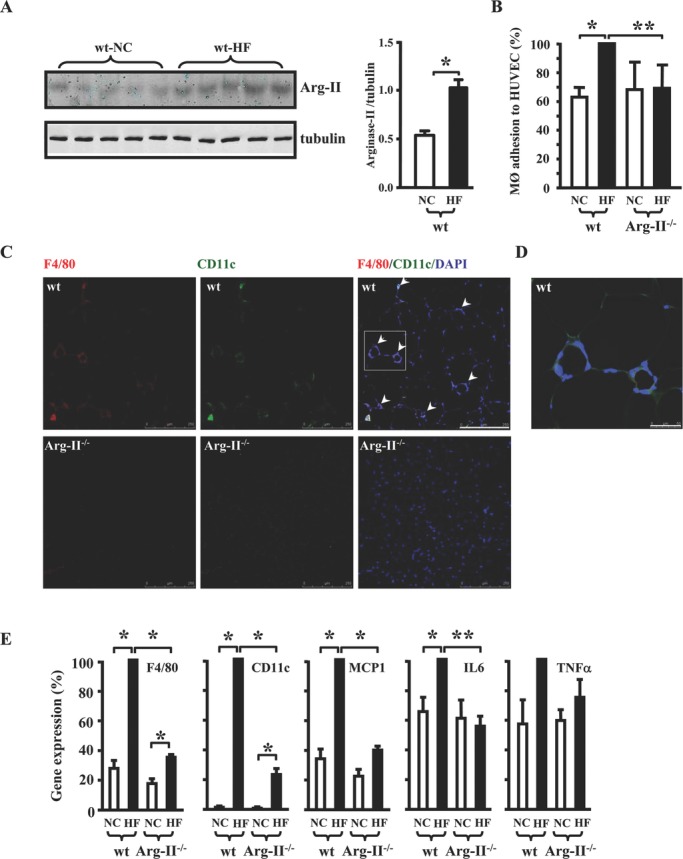
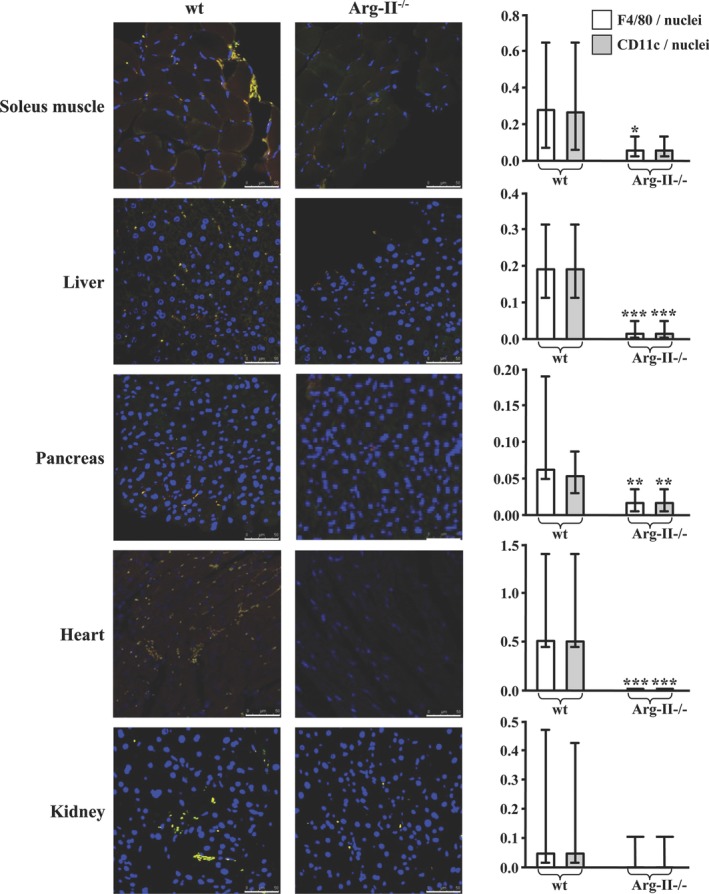
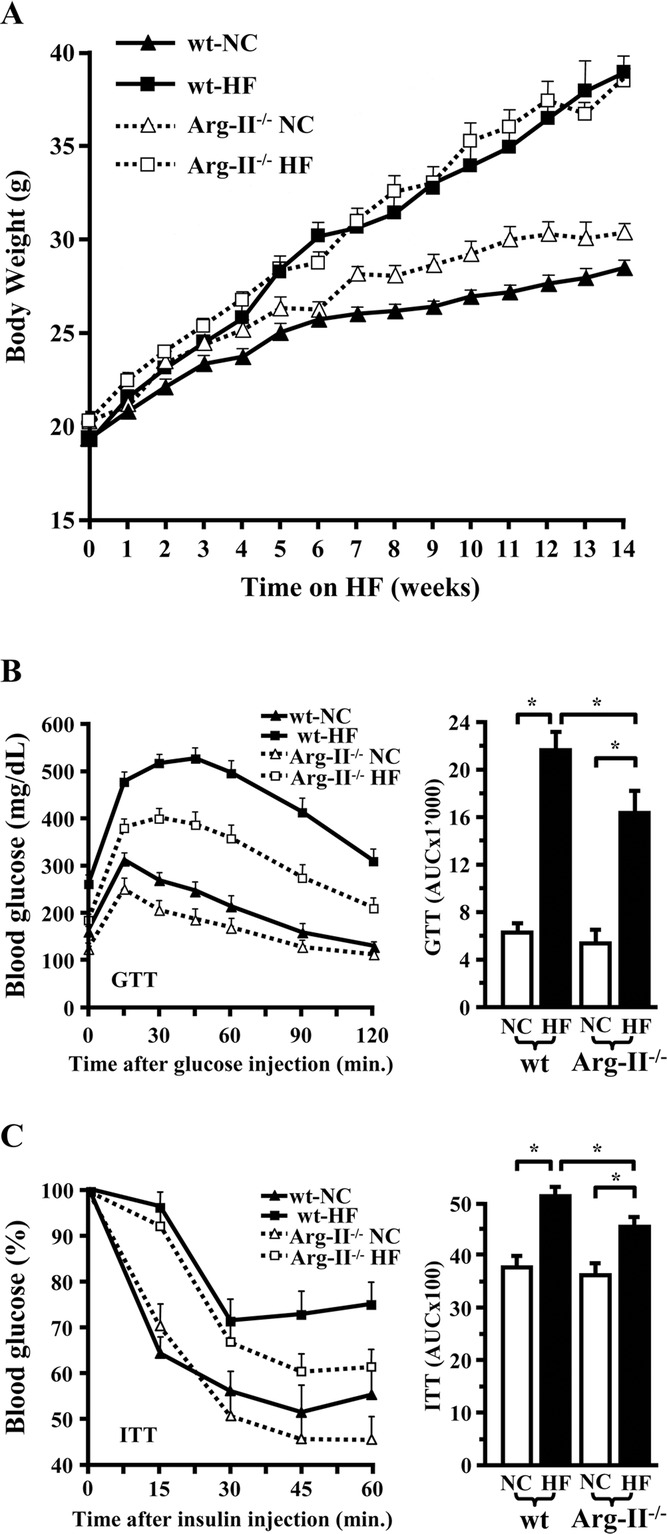
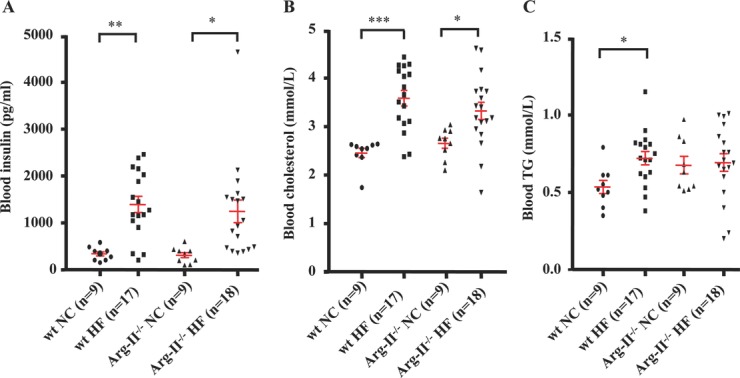
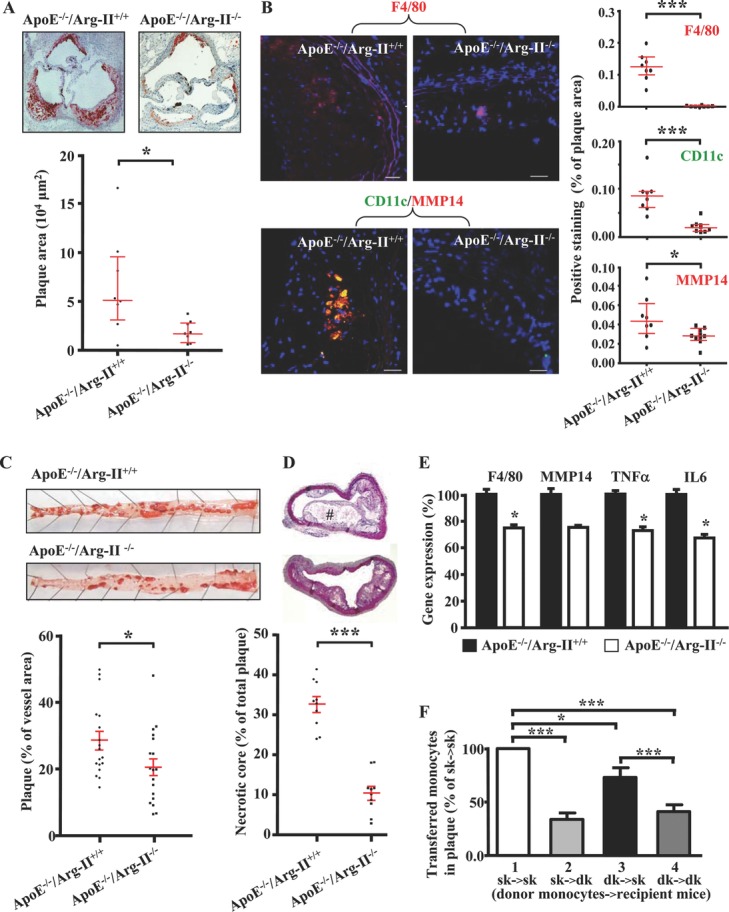
Methods and results: In human monocytes, silencing Arg-II decreases the monocytes' adhesion to endothelial cells and their production of proinflammatory mediators stimulated by oxidized low-density lipoprotein or lipopolysaccharides, as evaluated by real-time quantitative reverse transcription-polymerase chain reaction and enzyme-linked immunosorbent assay. Macrophages differentiated from bone marrow cells of Arg-II-deficient (Arg-II(-/-)) mice express lower levels of lipopolysaccharide-induced proinflammatory mediators than do macrophages of wild-type mice. Importantly, reintroducing Arg-II cDNA into Arg-II(-/-) macrophages restores the inflammatory responses, with concomitant enhancement of mitochondrial reactive oxygen species. Scavenging of reactive oxygen species by N-acetylcysteine prevents the Arg-II-mediated inflammatory responses. Moreover, high-fat diet-induced infiltration of macrophages in various organs and expression of proinflammatory cytokines in adipose tissue are blunted in Arg-II(-/-) mice. Accordingly, Arg-II(-/-) mice reveal lower fasting blood glucose and improved glucose tolerance and insulin sensitivity. Furthermore, apolipoprotein E (ApoE)-deficient mice with Arg-II deficiency (ApoE(-/-)Arg-II(-/-)) display reduced lesion size with characteristics of stable plaques, such as decreased macrophage inflammation and necrotic core. In vivo adoptive transfer experiments reveal that fewer donor ApoE(-/-)Arg-II(-/-) than ApoE(-/-)Arg-II(+/+) monocytes infiltrate into the plaque of ApoE(-/-)Arg-II(+/+) mice. Conversely, recipient ApoE(-/-)Arg-II(-/-) mice accumulate fewer donor monocytes than do recipient ApoE(-/-)Arg-II(+/+) animals.
Conclusions: Arg-II promotes macrophage proinflammatory responses through mitochondrial reactive oxygen species, contributing to insulin resistance and atherogenesis. Targeting Arg-II represents a potential therapeutic strategy in type II diabetes mellitus and atherosclerosis. (J Am Heart Assoc. 2012;1:e000992 doi: 10.1161/JAHA.112.000992.).
Keywords: atherosclerosis; diabetes mellitus, type 2; inflammation; macrophages.
Figures
Similar articles
-
MicroRNA-155 deficiency results in decreased macrophage inflammation and attenuated atherogenesis in apolipoprotein E-deficient mice.Arterioscler Thromb Vasc Biol. 2014 Apr;34(4):759-67. doi: 10.1161/ATVBAHA.113.302701. Epub 2014 Feb 6. Arterioscler Thromb Vasc Biol. 2014. PMID: 24504735 Free PMC article.
-
AMP-activated protein kinase α1 promotes atherogenesis by increasing monocyte-to-macrophage differentiation.J Biol Chem. 2017 May 12;292(19):7888-7903. doi: 10.1074/jbc.M117.779447. Epub 2017 Mar 22. J Biol Chem. 2017. PMID: 28330873 Free PMC article.
-
IgE Contributes to Atherosclerosis and Obesity by Affecting Macrophage Polarization, Macrophage Protein Network, and Foam Cell Formation.Arterioscler Thromb Vasc Biol. 2020 Mar;40(3):597-610. doi: 10.1161/ATVBAHA.119.313744. Epub 2020 Jan 30. Arterioscler Thromb Vasc Biol. 2020. PMID: 31996021 Free PMC article.
-
Protease-Activated Receptor-2 Plays a Critical Role in Vascular Inflammation and Atherosclerosis in Apolipoprotein E-Deficient Mice.Circulation. 2018 Oct 16;138(16):1706-1719. doi: 10.1161/CIRCULATIONAHA.118.033544. Circulation. 2018. PMID: 29700120
-
Macrophages, inflammation, and atherosclerosis.Int J Obes Relat Metab Disord. 2003 Dec;27 Suppl 3:S35-40. doi: 10.1038/sj.ijo.0802498. Int J Obes Relat Metab Disord. 2003. PMID: 14704742 Review.
Cited by
-
Arginase inhibition mediates renal tissue protection in diabetic nephropathy by a nitric oxide synthase 3-dependent mechanism.Kidney Int. 2013 Dec;84(6):1189-97. doi: 10.1038/ki.2013.215. Epub 2013 Jun 12. Kidney Int. 2013. PMID: 23760286 Free PMC article.
-
Arginase as a Critical Prooxidant Mediator in the Binomial Endothelial Dysfunction-Atherosclerosis.Oxid Med Cell Longev. 2015;2015:924860. doi: 10.1155/2015/924860. Epub 2015 May 4. Oxid Med Cell Longev. 2015. PMID: 26064427 Free PMC article. Review.
-
Hypoxia Enhances Endothelial Intercellular Adhesion Molecule 1 Protein Level Through Upregulation of Arginase Type II and Mitochondrial Oxidative Stress.Front Physiol. 2019 Aug 14;10:1003. doi: 10.3389/fphys.2019.01003. eCollection 2019. Front Physiol. 2019. PMID: 31474872 Free PMC article.
-
Role of Metabolites of Nitric Oxide and Arginase in the Pathogenesis of Glomerulonephritis.Curr Health Sci J. 2016 Jul-Sep;42(3):221-225. doi: 10.12865/CHSJ.42.03.01. Epub 2016 Sep 29. Curr Health Sci J. 2016. PMID: 30581575 Free PMC article.
-
Mechanisms of obesity-induced metabolic and vascular dysfunctions.Front Biosci (Landmark Ed). 2019 Mar 1;24(5):890-934. doi: 10.2741/4758. Front Biosci (Landmark Ed). 2019. PMID: 30844720 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
