

FOLD-EM: automated fold recognition in medium- and low-resolution (4-15 Å) electron density maps
- PMID: 23131460
- PMCID: PMC3519459
- DOI: 10.1093/bioinformatics/bts616
FOLD-EM: automated fold recognition in medium- and low-resolution (4-15 Å) electron density maps
Abstract
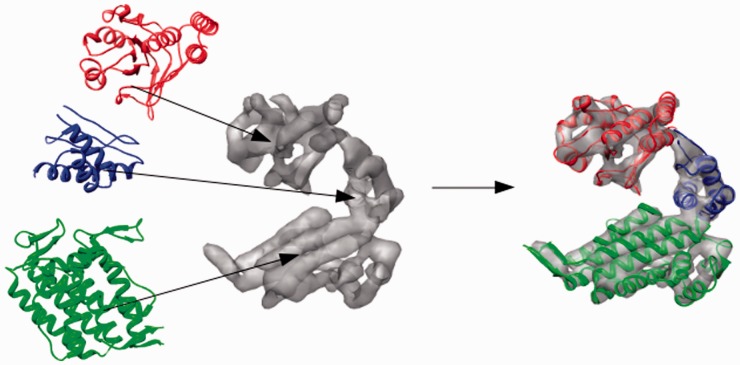
Motivation: Owing to the size and complexity of large multi-component biological assemblies, the most tractable approach to determining their atomic structure is often to fit high-resolution radiographic or nuclear magnetic resonance structures of isolated components into lower resolution electron density maps of the larger assembly obtained using cryo-electron microscopy (cryo-EM). This hybrid approach to structure determination requires that an atomic resolution structure of each component, or a suitable homolog, is available. If neither is available, then the amount of structural information regarding that component is limited by the resolution of the cryo-EM map. However, even if a suitable homolog cannot be identified using sequence analysis, a search for structural homologs should still be performed because structural homology often persists throughout evolution even when sequence homology is undetectable, As macromolecules can often be described as a collection of independently folded domains, one way of searching for structural homologs would be to systematically fit representative domain structures from a protein domain database into the medium/low resolution cryo-EM map and return the best fits. Taken together, the best fitting non-overlapping structures would constitute a 'mosaic' backbone model of the assembly that could aid map interpretation and illuminate biological function.
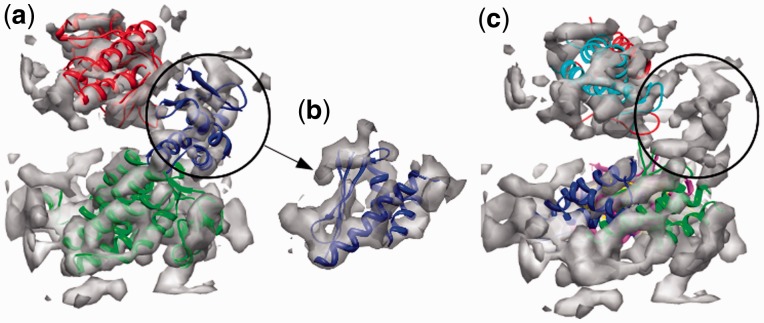
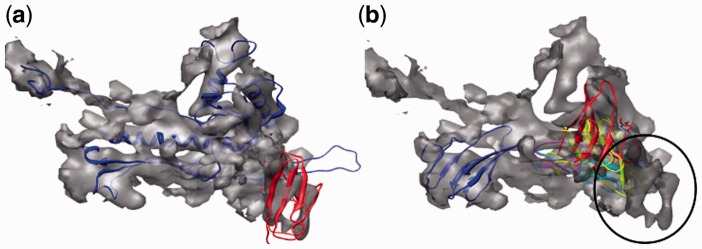
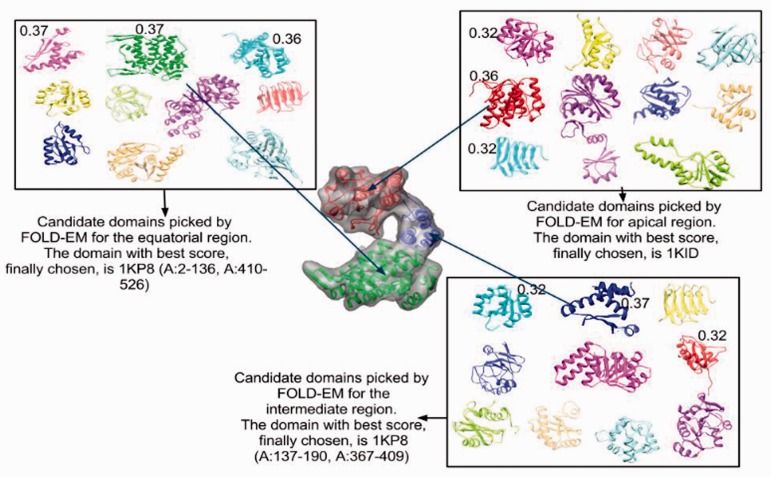
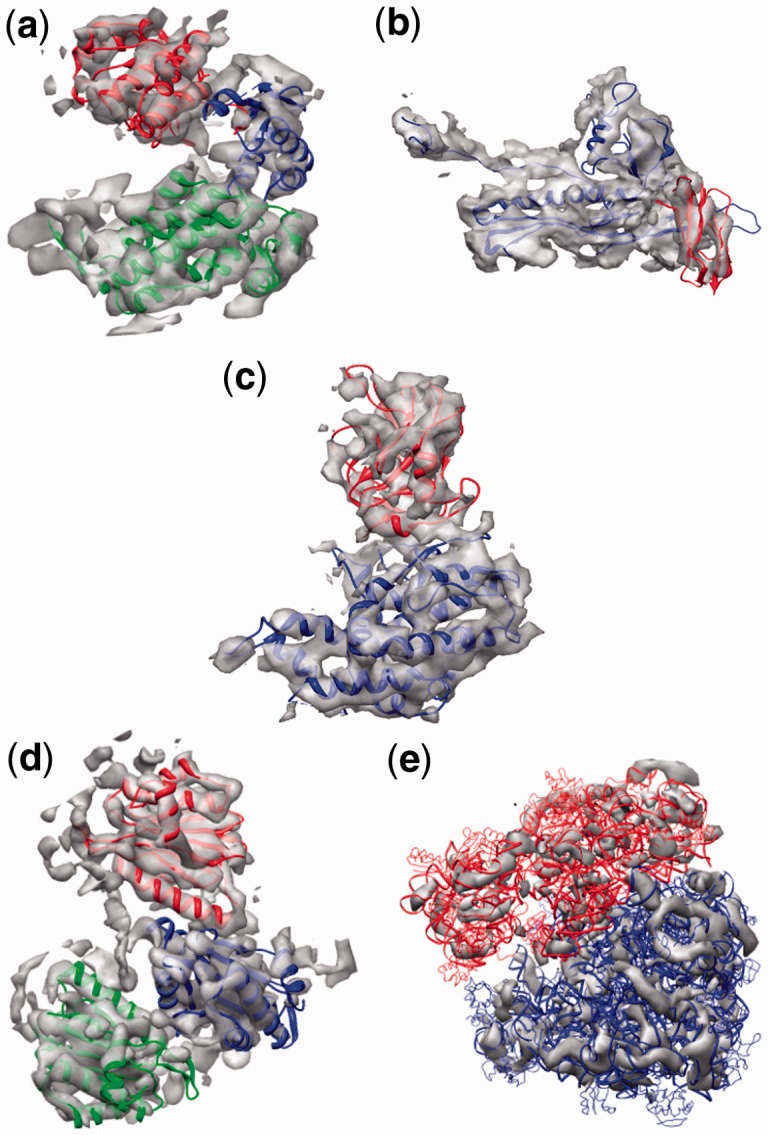
Result: Using the computational principles of the Scale-Invariant Feature Transform (SIFT), we have developed FOLD-EM-a computational tool that can identify folded macromolecular domains in medium to low resolution (4-15 Å) electron density maps and return a model of the constituent polypeptides in a fully automated fashion. As a by-product, FOLD-EM can also do flexible multi-domain fitting that may provide insight into conformational changes that occur in macromolecular assemblies.
Figures
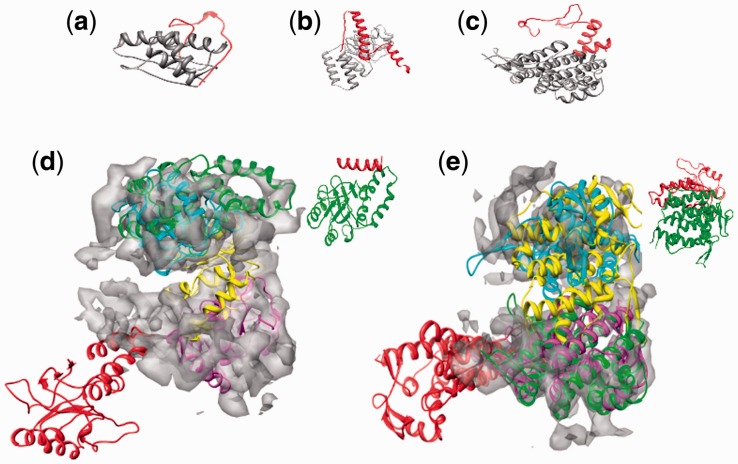
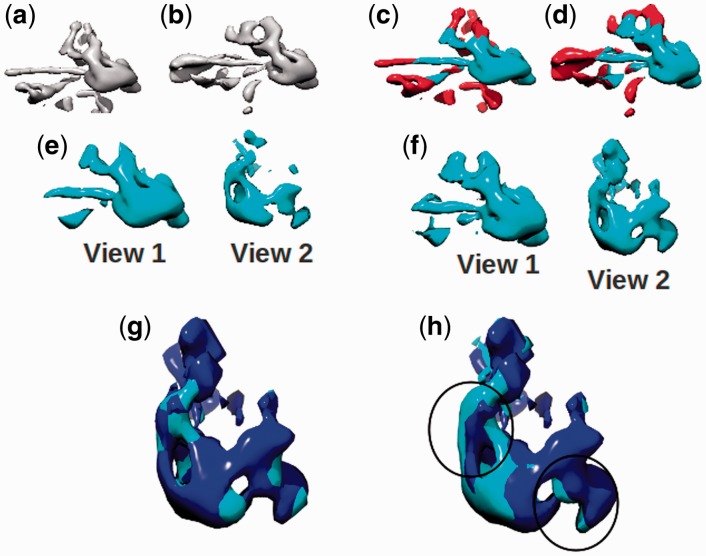
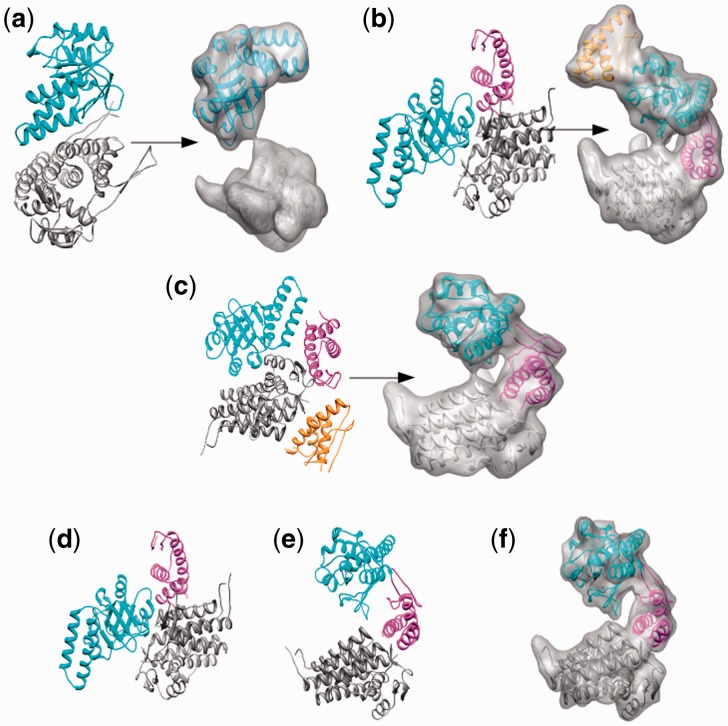
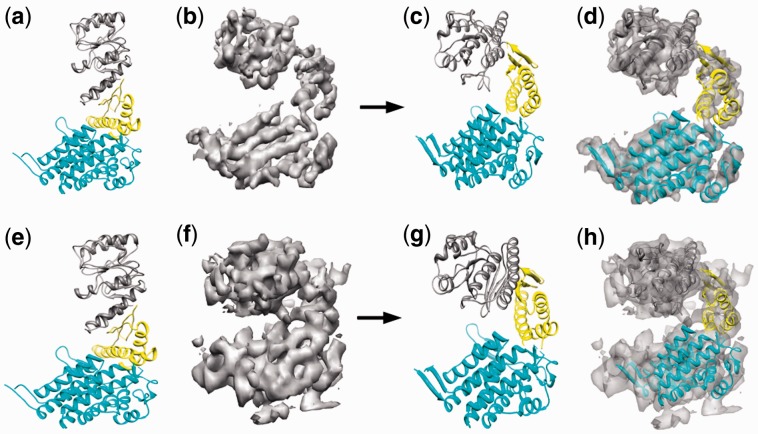
Similar articles
-
EMatch: an efficient method for aligning atomic resolution subunits into intermediate-resolution cryo-EM maps of large macromolecular assemblies.Acta Crystallogr D Biol Crystallogr. 2007 Jan;63(Pt 1):42-9. doi: 10.1107/S0907444906041059. Epub 2006 Dec 13. Acta Crystallogr D Biol Crystallogr. 2007. PMID: 17164525 Free PMC article.
-
EMatch: discovery of high resolution structural homologues of protein domains in intermediate resolution cryo-EM maps.IEEE/ACM Trans Comput Biol Bioinform. 2007 Jan-Mar;4(1):28-39. doi: 10.1109/TCBB.2007.1003. IEEE/ACM Trans Comput Biol Bioinform. 2007. PMID: 17277411
-
Protein-Protein Modeling Using Cryo-EM Restraints.Methods Mol Biol. 2020;2112:145-162. doi: 10.1007/978-1-0716-0270-6_11. Methods Mol Biol. 2020. PMID: 32006284
-
Progress in spatial resolution of structural analysis by cryo-EM.Microscopy (Oxf). 2023 Apr 6;72(2):135-143. doi: 10.1093/jmicro/dfac053. Microscopy (Oxf). 2023. PMID: 36269102 Review.
-
Refinement of Atomic Structures Against cryo-EM Maps.Methods Enzymol. 2016;579:277-305. doi: 10.1016/bs.mie.2016.05.033. Epub 2016 Jun 24. Methods Enzymol. 2016. PMID: 27572731 Review.
Cited by
-
Detecting protein and DNA/RNA structures in cryo-EM maps of intermediate resolution using deep learning.Nat Commun. 2021 Apr 16;12(1):2302. doi: 10.1038/s41467-021-22577-3. Nat Commun. 2021. PMID: 33863902 Free PMC article.
-
Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement.Nat Methods. 2015 Apr;12(4):361-365. doi: 10.1038/nmeth.3286. Epub 2015 Feb 23. Nat Methods. 2015. PMID: 25707030 Free PMC article.
-
Bayesian Weighing of Electron Cryo-Microscopy Data for Integrative Structural Modeling.Structure. 2019 Jan 2;27(1):175-188.e6. doi: 10.1016/j.str.2018.09.011. Epub 2018 Nov 1. Structure. 2019. PMID: 30393052 Free PMC article.
-
Integrative structural modeling of macromolecular complexes using Assembline.Nat Protoc. 2022 Jan;17(1):152-176. doi: 10.1038/s41596-021-00640-z. Epub 2021 Nov 29. Nat Protoc. 2022. PMID: 34845384 Review.
-
Improved metrics for comparing structures of macromolecular assemblies determined by 3D electron-microscopy.J Struct Biol. 2017 Jul;199(1):12-26. doi: 10.1016/j.jsb.2017.05.007. Epub 2017 May 25. J Struct Biol. 2017. PMID: 28552721 Free PMC article.
References
-
- Jiang W., et al. Bridging the information gap: computational tools for intermediate resolution structure interpretation. J. Mol. Biol. 2001;208:1033–1044. - PubMed
-
- Jiang W., et al. Coat protein fold and maturation transition of bacteriophage P22 seen at subnanometer resolutions. Nat. Struct. Biol. 2003;10:131–135. - PubMed
-
- Lasker K., et al. Discovery of protein substructures in EM maps. Algorithms in Bioinformatics. 2005;3692:423–434.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
